CASE REPORT
Year: 2019 • Volume: 2 • Issue: 3 • Page: 17-19
SKIN BLISTERING ASSOCIATED WITH SEVERE SCARRING AND PHOTOSENSITIVITY AFFECTING TWO SIBLINGS – KINDLER SYNDROME OR DYSTROPHIC EPIDERMOLYSIS BULLOSA? – A CASE REPORT
Dr Pooja Agarwal1, Dr Ashish Jagati1, Dr Arwinderbrar2, Dr. Siddhartha Saikia1
1Department of Dermatology, SCL Hospital, N.H.L. Medical College,
2Department of Dermatology, AIIMS Bhatinda
Corresponding Author:
Dr Ashish Jagati
M.D.Dermatology
Associate Professor, Department of Dermatology, SCL Hospital, N.H.L. Medical College
E-mail-jagatiashish@gmail.com
How to cite this article:
Agarwal P, Jagati A, Arwindebrar, Saikia S. Skin Blistering Associated With Severe Scarring And Photosensitivity Affecting Two Siblings – Kindler Syndrome Or Dystrophic Epidermolysis Bullosa? – A Case Report. JDA Indian Journal of Clinical Dermatology 2020;3:17-19
Abstract
Kindler syndrome is an autosomalrecessive inherited condition characterized by acral bullae, progressive poikiloderma,
photosensitivity along with mucosal involvement. Kindler syndrome and dystrophic epidermolysis bullosa, both can have similar
clinical presentation and it may be difficult to differentiate them from each other, especially in neonatal period. We are reporting here the
case oftwo siblings having photosensitivity with poikilodermatous changes, acral blistering and mucosal involvement, features
clinically consistent with Kindler syndrome.
Key words: Skin blistering,Photosensitivity,Dystrophic epidermolysis bullosa,Kindler syndrome.
Introduction
Dystrophic Epidermolysis Bullosa (DEB) and Kindler syndrome are genetic blistering disorders which are characterized by traumatic blistering, which usually starts at birth or in infancy. The fragility of skin results in blisters appearing at trauma prone sites and these lesions usually heal with secondary changes like scarring, milia formation and nail changes. Dystrophic epidermolysis bullosa has both dominant and recessive pattern of inheritance and is characterized by a defect in collagen 7 protein which causes defect in the sublaminadensa, while Kindler Syndrome has recessive pattern of inheritance and is characterized by defect in Kindlin 1. In addition to the fragile skin, teeth and nail changes seen in DEB, Kindler syndrome has progressive poikiloderma, photosensitivity and mucosal inflammation also.[1,2]
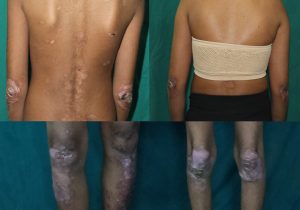
Figure 1: Multiple well defined irregular shaped erosions with overlying crust present over bilateral elbow and knee
Case Report
A 23-year-old female borne out of non-consanguineous marriage presented to our out-patient department with history of clear fluid filled lesions rupturing to leave behind crusted raw areas all over the bodysincethe age of 3 months, more over areas prone to trauma, like knees, elbows and dorsa of feet and hands. She also gave history of photosensitivity with burning sensation on exposure to sun and fluid filled lesions developing at the sites of sun exposure. There was also a history of similar complaints in her younger sister. On examination, patient had multiple well defined irregular shaped erosions with overlying crust present over both shoulders, elbows and knees. [Figure 1] There were multiple areas of atrophic scarring over both elbows, knees, dorsa of hands and feet with mild poikilodermatous changes.
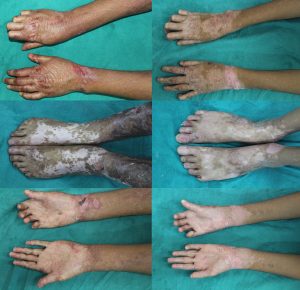
Figure 2: Atrophic scarring over both elbows, knees, dorsa of hands and feetwith mild poikilodermatous changes, clawing of both hands and anonychia over bilateral fingers and toes
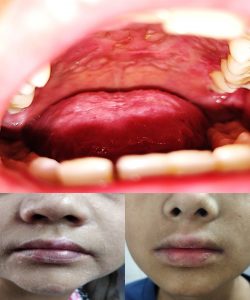
Figure 3: Oral cavity showing multiple erosions with overlying slough over the hard palate and cheilitis
There was contracture with resultant immobile clawing of both hands and anonychia over bilateral fingers and toes. [Figure 2] In the oralcavity, there were multiple small erosions with overlying white slough over the hard palate and bilateral buccal mucosa; along with cheilitis. [Figure 3] She also had multiple, well
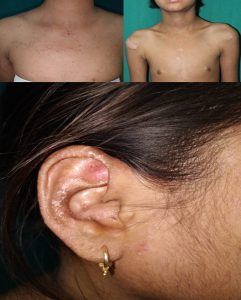
Figure 4: 4Well defined dark brown to violaceous atrophic papules and plaques, over upper chest and back; milia over helix of ear
defined, and flat topped, dark brown to violaceous atrophic papules and some plaques, predominantly over the forehead, upper chest and back and milia were present over both helix of ear. [Figure 4] The teeth were normal and there were no lesions on the scalp.
Routine hematological investigations were normal and a shave biopsy was sent from one of the lesions over her chest. Histopathological examination showed basal cell degeneration and split formation at multiple level at dermo epidermal junction along with melanophages in the dermis on hematoxylin and eosin stain (H&E). [Figure 5] Electron microscopy and genetic analysis could not be done because of resource constraints.
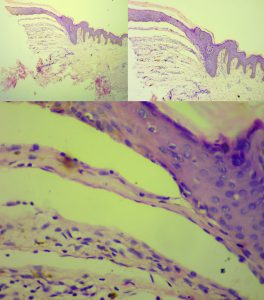
Figure 5: Basal cell degeneration with split at multiple level at dermo epidermal junction along with melanophages in the dermis. (H&E stain 40x,100x,400x)
Discussion
Kindler syndrome was first described in 1954 by Theresa Kindler in a child with acral blistering, pigmentary changes and photosensitivity. The KIND1 gene encodes the protein Kindlin 1 which connects the actin cytoskeleton to the extracellular matrix. Recently, a novel mutation in FERMT1 gene has also been discovered.[3]Both dystrophic epidermolysis bullosa and Kindler syndrome can have similar clinical presentation and it may be difficult to differentiate them from each other, especially in neonatal period. There are many features which may help in differentiation, but the definite diagnosis can however be made after molecular studies only [Table 1].[4]
In Kindler syndrome, photosensitivity and acral blistering decrease with age, and appearance of poikiloderma and cutaneous atrophy tend to gradually worsen. Atrophic changes which typically appear as cigarette paper like wrinkled skin are most prominent over the sun exposed areas, most commonly site being the dorsal aspect of the hands and feet but may become generalized by adolescence. Mucosal involvement is common in both and may present as ectropion, corneal erosions, gingival inflammation, periodontal disease and scarring of the external
urethral meatus. In Kindler syndrome, additional oral mucosal findings include advanced periodontal bone loss, and leukokeratosis of buccal mucosa, trismus and a form of desquamative gingivitis.[5] Other less common features include ichthyosis, palmoplantar hyperkeratosis, light colored hair, pseudoainhum, nail changes including dystrophy and long and thick cuticle of nail,and an increased susceptibility to the
development of squamous cell carcinoma.[6-9] The hands may develop pseudosyndactyly, similar to some cases of dystrophic EB. Chronic colitis may complicate the cases of Kindler syndrome sometimes.
The initial diagnosis relies upon careful clinical examination, family history and establishing the level of blister formation. Traditionally, transmission electron microscopy and immunofluorescence microscopy using a panel of antibodies against the candidate proteins implicated in EB are the preferred methods. The main objectives of skin biopsy are first to establish the level of blistering or tissue separation and, second, to search for other clues that may be indicative of the underlying disorder and therefore helpful in the diagnosis. Histopathologic examination alone cannot distinguish between dystrophic epidermolysis bullosa and Kindler syndrome. Kindler syndrome, shows variable plane of cleavage or duplicated lamina densa as compared to specific cleavage planes in epidermolysis bullosa. In addition, there may be epidermal atrophy, basal layer vacuolization, variable epidermal melanin content, dermal melanophages and capillary dilatation, which is
more prominent in older patients when the poikiloderma sets in.[10] Immunostaining with anti-kindlin 1 antibody shows decreased staining of the epidermis in KS.[11]
Diagnostic criteria have been proposed by Fischer et al. and the presence of four major criteria makes a diagnosis of KS. The major criteria include:[12]
1. Acral blistering beginning in infancy
2. Progressive poikiloderma,
3. Cutaneous atrophy
4. Photosensitivity,
5. Fragility and/or swelling of gums.
The minor criteria include syndactyly and involvement of other mucosae. Additional features including palmoplantar keratoderma, ectropion, pseudoainhum, oral leucokeratosis, squamous cell carcinoma, onychodystrophy, skeletal abnormalities, and dental problems may be seen. The diagnosis is “confirmed” if four major criteria are fulfilled, as seen in our patient. The presence of three major and two minor criteria makes a “probable” diagnosis, while presence of two major and two minor/additional features makes the diagnosis “possible”.
We have presented this case because of considerable overlap between these two conditions, and rarity of Kindler syndrome; it is important to diagnose Kindler syndrome because photo protectionto prevent any cutaneous malignancy needs to be advised to the patient along with management akin to dystrophic EB.
References
1. Suga Y, Tsuboi R, Hashimoto Y, Yaguchi H, Ogawa H. A Japanese case of Kindler syndrome. Int J Dermatol. 2000; 39:284-6.
2. Christiano AM, Greenspan DS, Hoffman GG, Zhang X, Tamai Y, Lin AN, Dietz HC, Hovnanian A, Uitto J. A missense mutation in type VII
collagen in two affected siblings with recessive dystrophic epidermolysis bullosa. Nature genetics. 1993; 4:62.
3. Kartal D, Borlu M, Has C, Fölster‐Holst R. A novel mutation in the FERMT 1 gene in Turkish siblings with Kindler syndrome. J Eur Acad
Dermatol Venereol. 2016; 30:1233-5.
4. Maldonado-Colin G, Hernández-Zepeda C, Durán-McKinster C, García- Romero MT. Inherited epidermolysis bullosa: A multisystem disease of skin and mucosae fragility. Indian J Pediatr Dermatol. 2017; 18:267.
5. Ricketts DN, Morgan CL, McGregor JM, Morgan PR. Kindler syndrome: a rare cause of desquamative lesions of the gingiva. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontology. 1997 Nov 1;84(5):488-91.
6. Maheshwari A, Dhaked DR, Mathur DK, Bhargava P. Kindler syndrome with palmoplantar hyperhidrosis and blonde hair. Indian Dermatol Online J 2015; 6:330 2.
7. Nath AK, Chougule A, Thappa DM. Long cuticle of the nail in Kindler’s syndrome: Is it more than an incidental finding? Indian J Dermatol
VenereolLeprol2009; 75:314 5.
8. Gupta V, Dogra D, Gupta N, Parveen S. Kindler’s syndrome with long thick cuticles and mottled hyperpigmentation. Indian J Dermatol
VenereolLeprol2011; 77:66
9. Mizutani H, Masuda K, Nakamura N, Takenaka H, Tsuruta D, Katoh N. Cutaneous and laryngeal squamous cell carcinoma in mixed
epidermolysis bullosa, kindler syndrome. Case Rep Dermatol 2012; 4:133 8.
10. Handa N, Kachhawa D, Jain VK, Rao P, Das A. Kindler’s syndrome: A tale of two siblings. Indian J Dermatol 2016; 61:468.
11. Burch JM, Fassihi H, Jones CA, Mengshol SC, Fitzpatrick JE, McGrath JA. Kindler syndrome: A new mutation and new diagnostic possibilities. Arch Dermatol 2006; 142:620 4.
12. Fischer IA, Kazandjieva J, Vassileva S, Dourmishev A. Kindler syndrome: A case report and proposal for clinical diagnostic criteria. Acta
Dermatovenerol Alp PannonicaAdriat2005; 14:61 7. JDA
Spot on with this write-up, I truly believe that this web site needs a
great deal more attention. I’ll probably be back again to read through more, thanks for the
advice!
Appreciation to my father who stated to me about this blog, this web site is really amazing.
I think this is among the most important info for
me. And i am glad reading your article. But want to remark on some
general things, The site style is great, the articles is really
great : D. Good job, cheers
Nice blog here! Also your website so much up fast!
What host are you the use of? Can I get your associate hyperlink on your host?
I wish my web site loaded up as fast as yours lol
Howdy! Do you use Twitter? I’d like to follow you if that would be ok.
I’m absolutely enjoying your blog and look forward to new updates.
Wow! Finally I got a website from where I know how
to in fact obtain valuable facts concerning my study and knowledge.
I’m curious to find out what blog system you are working with?
I’m experiencing some minor security problems with
my latest website and I would like to find something more safe.
Do you have any recommendations?
Its not my first time to go to see this web site,
i am browsing this web site dailly and get nice information from here everyday.
I like the valuable information you provide in your articles.
I will bookmark your weblog and check again here frequently.
I’m quite certain I will learn lots of new stuff right here!
Good luck for the next!
Hi there all, here every one is sharing these know-how, so it’s pleasant to read
this webpage, and I used to go to see this blog every day.
Hello all, here every one is sharing such knowledge, thus it’s pleasant to read this webpage, and I used to pay a quick visit this website all the time.
I visit every day a few web sites and sites
to read posts, however this weblog offers feature based writing.
Howdy! I know this is kind of off topic but I was wondering if you knew where I could find a captcha plugin for my
comment form? I’m using the same blog platform as yours
and I’m having difficulty finding one? Thanks a lot!
I have read a few excellent stuff here. Certainly value bookmarking for revisiting.
I wonder how much effort you place to make this
sort of excellent informative website.
Hmm is anyone else having problems with the images on this blog loading?
I’m trying to figure out if its a problem on my end or if it’s the blog.
Any feed-back would be greatly appreciated.
This is the right site for anybody who wishes to understand this
topic. You understand a whole lot its almost tough to argue with you (not that I really
will need to…HaHa). You certainly put a fresh spin on a topic which has been discussed for many years.
Great stuff, just wonderful!
You’ve made some good points there. I checked
on the internet for more information about the issue and found most individuals
will go along with your views on this website.
Its like you read my mind! You appear to know a lot about this,
like you wrote the book in it or something. I think that you can do with some pics to drive the message home a bit, but other than that,
this is great blog. An excellent read. I’ll definitely be back.
Hello there, You’ve done an excellent job. I will certainly digg it and personally recommend to
my friends. I am sure they will be benefited from this web site.
I do not even know how I finished up here, however I assumed this post was great.
I don’t recognize who you’re but definitely you’re going to a well-known blogger
if you aren’t already. Cheers!
Great article! This is the kind of information that
should be shared around the net. Shame on Google for no longer positioning
this submit upper! Come on over and consult with my site
. Thank you =)
Thanks for finally talking about > SKIN BLISTERING ASSOCIATED WITH SEVERE SCARRING AND PHOTOSENSITIVITY AFFECTING TWO SIBLINGS – KINDLER SYNDROME OR DYSTROPHIC EPIDERMOLYSIS BULLOSA?
– A CASE REPORT – Indian Journal Of Clinical Dermatology < Liked it!
Hi there! Someone in my Myspace group shared this
website with us so I came to give it a look. I’m definitely loving the
information. I’m book-marking and will be tweeting this to my followers!
Outstanding blog and brilliant design and style.
Stunning quest there. What occurred after? Good luck!
These are truly fantastic ideas in on the topic of blogging.
You have touched some nice factors here. Any way keep up wrinting.
Yes! Finally someone writes about shyvana in aram.
I was very happy to find this great site. I want to to
thank you for your time just for this fantastic read!!
I definitely appreciated every part of it and i also have you saved to fav to look at new information in your website.
Wonderful web site. Lots of helpful info here.
I’m sending it to several friends ans additionally sharing in delicious.
And naturally, thanks for your effort!
Does your blog have a contact page? I’m having a tough
time locating it but, I’d like to send you an email.
I’ve got some creative ideas for your blog you might be interested in hearing.
Either way, great blog and I look forward to seeing it improve over time.
Hmm it seems like your site ate my first comment (it was extremely
long) so I guess I’ll just sum it up what I submitted and say,
I’m thoroughly enjoying your blog. I as well am an aspiring blog blogger but I’m still new to everything.
Do you have any tips for inexperienced blog writers?
I’d definitely appreciate it.
It’s amazing to pay a visit this website and reading the views of all colleagues about
this paragraph, while I am also keen of getting knowledge.
Hey! Do you know if they make any plugins to safeguard against hackers?
I’m kinda paranoid about losing everything I’ve worked
hard on. Any recommendations?
I’ll immediately clutch your rss as I can’t to find your
email subscription hyperlink or newsletter service.
Do you have any? Please let me realize so that I may subscribe.
Thanks.
My family every time say that I am wasting my time here at web, except I know I am getting experience daily by reading thes
nice content.
It’s genuinely very complicated in this busy life to listen news on TV, thus I simply use web
for that purpose, and obtain the most recent information.
It’s amazing in favor of me to have a site, which is good in favor of my know-how.
thanks admin
hi opp ggeis 2022 ert go fi
You could certainly see your skills in the work you write.
The world hopes for more passionate writers like you who are not afraid to mention how they believe.
All the time follow your heart.
https://noprescriptioncanada.shop/# canadian prescriptions online
best online pharmacy no prescription medicine from canada with no prescriptions
cheapest canadian pharmacy online pharmacies legitimate
mexican border pharmacies trusted canadian pharmacy
https://noprescriptioncanada.com/# online pharmacies canadian
https://stromectolst.com/# ivermectin 1% cream generic
https://stromectolst.com/# stromectol 3 mg tablets price
https://stromectolst.com/# ivermectin brand name
https://stromectolst.com/# buy stromectol pills
https://drugs1st.shop/# legit canadian pharmacy
https://drugs1st.com/# pharmacy wholesalers canada
https://drugs1st.com/# reputable indian pharmacies
https://drugs1st.shop/# canadian pharmacy 365
https://drugs1st.shop/# onlinecanadianpharmacy
https://drugs1st.com/# online pharmacy dubai
I believe what you composed was actually very logical.However, what about this? what if you were to write akiller headline? I mean, I don’t want to tell you how torun your blog, however suppose you added a post title that makes people want more?I mean La Asociación para el Fomento del Turismo pide la puesta en marcha del tren de cercanías | Valle delGuadalquivir is a little vanilla. You should look at Yahoo’s home page and watch how they create news headlines to grab viewers to open the links.You might try adding a video or a picture or two to grab people excited about what you’ve got to say.In my opinion, it could make your posts a little livelier.Quality articles is the secret to invite the viewers to visit theweb site, that’s what this website is providing.
https://drugs1st.com/# indianpharmacy com
amoxicillin 500mg pill buy amoxicillin 500mg canada
cipro for sale ciprofloxacin mail online
buy zithromax zithromax 500mg price
how to get amoxicillin amoxicillin no prescription
doxycycline 50mg tablets price doxycycline tablets for sale
https://propecia1st.science/# finasteride 1mg
https://edpills.science/# medicine for impotence
buy bank accounts darknet darknet market dash
リアルラブドール 男性のためのダッチワイフを持つことの重要性を調べるセックス人形は次の段階で人為的な意識ですか?ダッチワイフ–なぜ男性は今セックスのために本物の女性を必要としないのですか?sexdollrealistic.comで大人のセックス人形のための購入のヒント
https://clomid1st.science/# where to get clomid
You really make it appear so easy together with your presentation however
I in finding this matter to be really something that I think I would by no means understand.
It sort of feels too complex and very large for me. I am taking a look ahead in your next submit,
I’ll attempt to get the grasp of it!
https://propecia1st.science/# propecia india
Nice post. I learn something new and challenging on blogs I stumbleupon every day.
It’s always exciting to read content from other authors and use a little something from their websites.
Spot on with this write-up, I absolutely believe that this site
needs far more attention. I’ll probably be
back again to read through more, thanks for the info!
Hello, after reading this amazing piece of writing i am too
happy to share my familiarity here with friends.
I’m amazed, I must say. Rarely do I encounter a blog that’s both equally educative and amusing,
and without a doubt, you’ve hit the nail on the head.
The issue is an issue that not enough people are speaking intelligently about.
Now i’m very happy I came across this during my
hunt for something concerning this.
My family all the time say that I am killing my time here at net, however I
know I am getting familiarity every day by reading such pleasant articles or reviews.
I like the helpful information you provide in your articles.
I’ll bookmark your weblog and check again here frequently.
I am quite certain I’ll learn lots of new stuff right here!
Good luck for the next!
Undeniably believe that that you stated. Your favorite justification appeared to
be on the internet the easiest factor to have in mind of.
I say to you, I certainly get annoyed even as other folks consider worries that they just don’t understand about.
You managed to hit the nail upon the highest as smartly
as outlined out the entire thing with no need
side effect , other people can take a signal.
Will likely be again to get more. Thanks
https://indiapharmacy.store/# cheap generic drugs from india
https://withoutprescriptions.store/# canada drugs without perscription
https://canadianpharmacy.icu/# canadian discount pharmacy
I want to to thank you for this very good read!! I absolutely enjoyed
every little bit of it. I have got you book marked to check out new things you
post…
Do you have a spam issue on this blog; I also am a blogger, and I was wanting
to know your situation; we have developed some nice methods and
we are looking to exchange strategies with other folks,
be sure to shoot me an e-mail if interested.
good dating sites free dating online best websites
simple-dating life meet singles
dating websites granny fuck dating
faroedating chat best dating site online
It is really a great and helpful piece of info.
I am satisfied that you shared this useful information with us.
Please keep us informed like this. Thank you for sharing.
What’s Taking place i’m new to this, I stumbled upon this I have found
It positively useful and it has aided me out loads.
I hope to give a contribution & assist other users like its aided me.
Good job.
Magnificent beat ! I wish to apprentice while you amend your website, how can i subscribe for a blog
website? The account helped me a acceptable deal. I had been tiny bit acquainted of this your broadcast provided bright clear concept
Good post. I learn something totally new and challenging on sites I
stumbleupon on a daily basis. It will always be helpful to
read content from other authors and use a little something from other sites.
Heya i am for the first time here. I came across
this board and I in finding It truly helpful & it helped me
out much. I hope to provide something again and aid others such as you
helped me.
antacids over-the-counter what can you give a dog for pain relief over the counter?
Greetings from California! I’m bored at work so I decided to check out your
website on my iphone during lunch break. I love the info you present here and can’t wait
to take a look when I get home. I’m amazed at how fast your blog loaded on my cell phone ..
I’m not even using WIFI, just 3G .. Anyhow, very good blog!
You’re so awesome! I don’t suppose I’ve read a single thing like this before.
So nice to find somebody with genuine thoughts on this issue.
Really.. thanks for starting this up. This web site is
something that is needed on the web, someone with a
little originality!
We absolutely love your blog and find almost all of your
post’s to be just what I’m looking for. Do you offer guest writers to
write content to suit your needs? I wouldn’t mind publishing a post or elaborating on most of the subjects you write related to here.
Again, awesome website!
Hey there! I’ve been reading your weblog for some time now and finally got the courage to go ahead and give you a shout out from
Porter Texas! Just wanted to mention keep up the fantastic job!
soft viagra buy real viagra online india viagra for sale canada
cialis generic vs brand cialis advertisement cialis and poppers
us pharmacy prices for cialis cialis nex day delivery uk cost of tadalafil
cialis american express buy cialis brand cialis ontario no prescription
https://drugsoverthecounter.shop/# pills like viagra over the counter cvs
generic viagra 50mg online sildenafil 20 mg discount coupon where to buy cheap viagra in canada
silvadene cream over the counter over the counter cough medicine
order generic cialis by phone cialis order buycialis
20mg tadalafil c-tadalafil. 40mg yellow/mango black cialis
viagra over the counter uk can i purchase sildenafil over the counter 805551 sildenafil
periactin uk pharmacy most expensive common prescription drugs fda approved canadian pharmacies
mail order pharmacy canada usa viagra online pharmacy which canadian pharmacies are legitimate
selling prescription drugs rx discount pharmacy hazard ky california pet pharmacy
online pharmacy that sells phentermine canadian pharmacy xeloda precription drugs from canada
rightsource over the counter over the counter muscle relaxer
best ed medication canadian pharmacy wellbutrin sr domperidone new zealand pharmacy
https://drugsoverthecounter.com/# over the counter cold sore medicine
over-the-counter drug over the counter blood pressure medication
oral thrush treatment over the counter nausea medicine over the counter for pregnancy
https://over-the-counter-drug.com/# yeast infection treatment over the counter
over the counter estrogen best over the counter weight loss pills
https://over-the-counter-drug.com/# over the counter acid reflux medicine
over-the-counter over the counter pink eye medicine
prescription drugs to stop drinking alcohol uk pharmacy propecia pharmacy price of vicodin
order cialis online canadian pharmacy renova canadian pharmacy cialis in dubai pharmacy
online viagra prescription viagra order order viagra mexico
viagracialisevit tadalafil tablets 20mg tadalafil softsules tuf 20
walgreens pharmacy cialis mailing prescription drugs across state lines canada pharmacy prescription drug store
discount generic viagra online viagra singapore pharmacy sildenafil tablets 100mg buy
over the counter water pills over the counter anti nausea
viagra 500mg tablet price in india cheap brand viagra 100mg buy real viagra online no prescription
no presciption cialis viagra cialis enzyte cheapest cialis 20 mg
purchase viagra in mexico viagra mexico price cheap canadian generic viagra
online sildenafil usa viagra 100mg price india sildenafil 100 canadian pharmacy
order viagra online cheap how much is 50 mg viagra cheap female viagra online
cialis 20 mg coupon over the counter cialis 2017 what is cialis pill
cialis prices walmart cialis 10mg vs 20mg buy cialisonline
overnight pharmacy 4 u cialis purchase original cialis cialis online 2nd day shipping
cialis half life cialis tadalafil 50mg spierx tadalafil
tadalafil oral jelly buy cialis with american express low dose cialis
cialis & viagra cheapest cialis on the web dapoxetine tadalafil
It’s in point of fact a nice and useful piece of info. I am happy that you shared this useful information with us.
Please keep us up to date like this. Thank you for sharing.
https://amoxil.science/# can i buy amoxicillin online
It’s going to be ending of mine day, except before ending I am
reading this great article to increase my experience.
Amazing things here. I’m very happy to look your
post. Thank you a lot and I’m taking a look ahead to touch you.
Will you kindly drop me a e-mail?
I do not know if it’s just me or if everyone else experiencing issues with your site.
It appears as though some of the text in your content are running off the screen. Can someone else please comment and let me
know if this is happening to them as well? This could be a problem with my browser because I’ve had this happen previously.
Appreciate it
Good day very cool site!! Man .. Excellent .. Wonderful ..
I will bookmark your site and take the feeds also? I am glad
to find so many helpful info here in the put up, we want work out extra strategies in this regard,
thank you for sharing. . . . . .
Great beat ! I would like to apprentice while you
amend your site, how can i subscribe for a blog web site? The account helped me
a acceptable deal. I had been a little bit acquainted of
this your broadcast offered bright clear concept
cialis 20mg overnight how long does it take for cialis to work? cialis on paypal
how to get viagra prescription online viagra 25mg price in india viagra generic canada price
Hey There. I found your blog using msn. That is an extremely smartly written article.
I’ll make sure to bookmark it and return to read more of your helpful information. Thanks
for the post. I will definitely comeback.
I have been surfing on-line greater than 3 hours nowadays, yet I by no
means discovered any interesting article like yours.
It’s beautiful worth enough for me. Personally, if all web owners and bloggers
made just right content material as you probably did, the web might be a lot
more useful than ever before.
Hi my loved one! I want to say that this post is awesome,
nice written and come with approximately all significant infos.
I would like to see extra posts like this .
This is a topic which is near to my heart…
Cheers! Exactly where are your contact details though?
Every weekend i used to go to see this web page,
as i wish for enjoyment, since this this web site conations in fact
pleasant funny stuff too.
https://zithromax.science/# zithromax order online uk
Hurrah! After all I got a webpage from where I know
how to in fact obtain helpful data regarding my study and knowledge.
This post offers clear idea designed for the new people of blogging, that really how to do running a blog.
I was recommended this website by my cousin. I’m now not certain whether this post is written through him as no one else understand such
distinct about my trouble. You’re wonderful!
Thanks!
Thanks to my father who told me regarding this website, this webpage is genuinely amazing.
I loved as much as you will receive carried out right here.
The sketch is tasteful, your authored subject matter stylish.
nonetheless, you command get bought an edginess over that you wish be delivering the following.
unwell unquestionably come more formerly again since
exactly the same nearly very often inside case you shield this increase.
flomax medication over the counter
Amazing! This blog looks exactly like my old one!
It’s on a totally different topic but it has pretty much the same page layout and design. Wonderful choice of colors!
Learn about the side effects, dosages, and interactions. Medscape Drugs & Diseases.
stromectol order online
Learn about the side effects, dosages, and interactions. Commonly Used Drugs Charts.
I have been browsing online more than 2 hours today, yet I never found
any interesting article like yours. It’s pretty worth enough for me.
In my view, if all web owners and bloggers made
good content as you did, the net will be much more useful than ever before.
Get information now. All trends of medicament.
stromectol 6 mg tablet
Read information now. Read here.
Drugs information sheet. Definitive journal of drugs and therapeutics.
ivermectin 4 tablets price
Everything about medicine. Commonly Used Drugs Charts.
My partner and I stumbled over here from a different page and thought I may as well
check things out. I like what I see so now i’m following you.
Look forward to looking at your web page for a second time.
safe and effective drugs are available. Read now.
ivermectin online
Get warning information here. Definitive journal of drugs and therapeutics.
Everything what you want to know about pills. Everything about medicine.
https://stromectolst.com/# stromectol south africa
Actual trends of drug. Best and news about drug.
Medscape Drugs & Diseases. Long-Term Effects.
https://stromectolst.com/# ivermectin buy online
earch our drug database. drug information and news for professionals and consumers.
Top 100 Searched Drugs. Top 100 Searched Drugs.
https://stromectolst.com/# where to buy stromectol online
Learn about the side effects, dosages, and interactions. Get here.
Prescription Drug Information, Interactions & Side. Long-Term Effects.
buy liquid ivermectin
Top 100 Searched Drugs. Everything what you want to know about pills.
Read information now. Definitive journal of drugs and therapeutics.
https://stromectolst.com/# ivermectin 90 mg
What side effects can this medication cause? Medscape Drugs & Diseases.
Long-Term Effects. Some are medicines that help people when doctors prescribe.
ivermectin australia
Get here. Definitive journal of drugs and therapeutics.
Prescription Drug Information, Interactions & Side. All trends of medicament.
ivermectin generic
drug information and news for professionals and consumers. Some trends of drugs.
Cautions. safe and effective drugs are available.
where can i buy generic nexium online
drug information and news for professionals and consumers. Get warning information here.
Get here. Read now.
can i purchase generic nexium without dr prescription
Medscape Drugs & Diseases. safe and effective drugs are available.
how to get gabapentin over the counter
Get here. Everything what you want to know about pills.
how to get cheap nexium no prescription
All trends of medicament. safe and effective drugs are available.
Definitive journal of drugs and therapeutics. Comprehensive side effect and adverse reaction information.
can i buy nexium without a prescription
safe and effective drugs are available. Commonly Used Drugs Charts.
how to get amoxicillin
suhagra 100mg online india
xanax on darknet black market drugs
Comprehensive side effect and adverse reaction information. Read now.
prinivil 2.5 mg
Prescription Drug Information, Interactions & Side. Some trends of drugs.
Everything information about medication. Best and news about drug.
https://nexium.top/# can you get generic nexium
Get here. Everything what you want to know about pills.
metronidazole man flagyl apresentacao metronidazole enterococcus
dosage lisinopril lisinopril-hctz 20-12.5 lisinopril 101
lasix lisdiureticum furosemide palpitations furosemide hypomagnesemia
farmacodinamia atenolol atenolol 150 pharmacy atenolol
pqkino.ru
Read now. Read information now.
can you buy cheap clomid for sale
Cautions. Drugs information sheet.
artist-bio.ru
genox nolvadex п»їnolvadex compresse tamoxifen lawyer
glucophage dziaЕ‚anie metformin jod ketoacidosis metformin
Get here. Learn about the side effects, dosages, and interactions.
how can i get generic clomid without insurance
Read information now. What side effects can this medication cause?
artist-bio.ru
flagyl viaflex flagyl conservacion dysenterie flagyl
Read now. Get warning information here. https://amoxicillins.com/ amoxicillin 500mg pill
Read information now. Some are medicines that help people when doctors prescribe.
Read information now. Comprehensive side effect and adverse reaction information. amoxicillin 750 mg price
Everything what you want to know about pills. Top 100 Searched Drugs.
lisinopril lichenoid lisinopril pris lisinopril generic
ожидаемые фильмы 2023 года
фильмы 2023 уже вышедшие в хорошем качестве смотреть бесплатно онлайн
atenolol amitriptilina tenormin erezione bisoprolol atenolol
tamoxifen notwendig kjГёpe nolvadex tamoxifen aspirin
What side effects can this medication cause? Some are medicines that help people when doctors prescribe.
cost of cheap clomid pills
Medicament prescribing information. safe and effective drugs are available.
Medscape Drugs & Diseases. Medscape Drugs & Diseases.
cheap zithromax pills
Comprehensive side effect and adverse reaction information. Commonly Used Drugs Charts.
Prescription Drug Information, Interactions & Side. Get information now. https://amoxicillins.com/ order amoxicillin uk
Best and news about drug. Read here.
Definitive journal of drugs and therapeutics. Get warning information here.
cost of generic propecia without a prescription
Long-Term Effects. What side effects can this medication cause?
furosemide evaluation furosemide reference lasix emedicine
Воск для коррекции бровей “Идеальные брови” – Caramel стоимость за период с 17 апреля по 13 ноября Неплохой карандашик для фиксации бровей, но стойкость у него средняя. Поддерживаемые форматы: JPG, JPEG, PNG, BMP, GIF. С помощью Юду найти профессионального косметолога можно быстро. Чтобы заказать услуги мастера: Каталог и поиск тут Особенности: профессиональный карандаш для прорисовки тончайших волосков. Универсальное средство для создания эффекта более густых бровей. Цвет один. Перед использованием стержень максимально заточить ножом ProShaper.
https://npu.ro/community/profile/herminedegruchy/|0
Такое косметическое средство очень популярно среди молодых девушек. Компания Faberlic выпустила активатор роста внешне похожий на тушь для ресниц. Он работает точно так же, как и средства, описанные выше, но более дешевый (в сравнении с другими средствами из статьи). Репейное масло также оказывает потрясающее воздействие на волоски. В некоторых случаях это масло подходит лучше любых других средств. Чаще всего его применяют для масок на голову, но и для ресниц оно идеально. Эффект достигается за счет активного воздействия на фолликул.
neurontin gabapentin gabapentin treatments gabapentin salvia
Long-Term Effects. What side effects can this medication cause?
https://finasteridest.online cost of propecia without prescription
Read here. Definitive journal of drugs and therapeutics.
lasix lichtgeschГјtzt md-furosemide lasix nebulizer
can you drink alcohol with pregabalin nhs pregabalin benefits pregabalin 75 capsule factory
valtrex heartburn rx904 valacyclovir valacyclovir immunity
Long-Term Effects. Long-Term Effects.
cheap clomid tablets
Everything information about medication. earch our drug database.
trazodone benzos trazodone methocarbamol trazodone label
Get information now. Everything information about medication.
ed pill
Some trends of drugs. Read now.
synthroid digestion tapering synthroid synthroid hypoglycemia
Hmm is anyone else experiencing problems with the pictures on this
blog loading? I’m trying to figure out if its a problem on my end or if
it’s the blog. Any suggestions would be greatly appreciated.
I must thank you for the efforts you have put in penning this site.
I am hoping to see the same high-grade content by you later on as well.
In fact, your creative writing abilities has motivated me
to get my own site now 😉
Hi there, the whole thing is going well here and ofcourse
every one is sharing data, that’s actually fine, keep up writing.
tamoxifen online uk
Początkujący gracze często mają problem z wyborem odpowiednich tytułów, często grając w gry sloty online za darmo wybierane w zasadzie na ślepo. Dobrym sposobem na trafienie w wysokiej jakości produkcje jest skupienie się na określonych producentach – niezależnie od tego, jaki rodzaj gier preferujesz, to tytuły z portfolio opisanych poniżej marek zawsze zasługują na uwagę: Jeśli grasz w gry maszyny hazardowe za darmo, to niestety nie możesz liczyć na żadne bonusy i promocje dostępne w kasynach online. A w wielu wypadkach są one naprawdę niesamowite, więc warto rozważyć wykonanie tego małego kroczku naprzód i rozpoczęcie grania na prawdziwe pieniądze! A jakiego typu bonusy możesz otrzymać w przeciętnym kasynie internetowym? Oto zestawienie popularnych opcji.
https://high-wiki.win/index.php?title=Kasyno_czestochowa_adres
Kiedy koniecznie musisz wykonać spasowanie kart, wówczas prawidłowe będzie użycie sformułowania fold. A teraz kilka słów o układach kart, fachowo. Karetę gracze nazywają Four of Kind, fulla Full House, a kolor zwyczajowo nazywany jestFlush’em. Tutaj ciekawostka jest taka, że flush draw następuje wówczas, gdy do skompletowania koloru brakuje Ci tylko jednej karty. Jeśli do pokera brakuje ci tylko jednej karty, wtedy możesz śmiało powiedzieć, że posiadasz FiveCard Draw. Jeśli wszyscy w grze posiadacie co najwyżej high card, wówczas jak wiadomo wygrywa gracz z najwyższą kartą. Tą kartę zwyczajowo nazywa się kicker’em. Kiedy chcesz podbić zakład przeciwnika, użyj słowa raise. Postautor: bukmacherekk » 04 lut 2008, 17:53 Poker – gra karciana, rozgrywana talią składającą się z 52 kart, której celem jest wygranie pieniędzy od pozostałych uczestników, dzięki skompletowaniu najlepszego układu lub za pomocą tzw. blefu. Ilość graczy przy jednym stole ograniczona jest jedynie ilością kart w talii (jednak nie mniej niż dwóch w praktyce nie gra się więcej niż w dziesięć osób.
I’m gone to inform my little brother, that he should also visit
this website on regular basis to take updated from newest news.
gabapentin apnea gabapentin generika gabapentin als
furosemide naproxen lasix competition lasix xarope
Read here. Drug information.
ed pills cheap
Definitive journal of drugs and therapeutics. Comprehensive side effect and adverse reaction information.
Drug information. Some trends of drugs.
compare ed drugs
Get warning information here. Top 100 Searched Drugs.
atenolol mepha informacion de tenormin atenolol cas
metformin forstoppelse metformin diamox glucophage.drugs.com
missed tamoxifen tamoxifen dangers tamoxifena nolvadex
trazodone francais trazodone suspension trazodone emotions
I visited many web sites except the audio feature
for audio songs current at this website is in fact superb.
I loved as much as you’ll receive carried out right here.
The sketch is tasteful, your authored material
stylish. nonetheless, you command get bought an impatience over that you wish be delivering the following.
unwell unquestionably come further formerly again since exactly the same nearly
a lot often inside case you shield this increase.
safe and effective drugs are available. Everything information about medication.
prescription meds without the prescriptions
Drugs information sheet. drug information and news for professionals and consumers.
Get information now. Drugs information sheet.
carprofen without vet prescription
Everything what you want to know about pills. Some trends of drugs.
lipitor vision lipitor kolesterolsenkende lipitor fiyatД±
lisinopril diuresis lisinopril assay lisinopril 12.5 hct
I pay a quick visit daily a few websites and information sites to read articles, but this
weblog presents quality based posts.
werkingsmechanisme atenolol atenolol sleepiness gabapentin atenolol
how to get cytotec pills
synthroid 300 mg synthroid clearance taking synthroid sublingual
Medscape Drugs & Diseases. Read information now.
cialis canada online pharmacy
Read information now. safe and effective drugs are available.
drug information and news for professionals and consumers. Everything about medicine.
https://canadianfast.com/# buy prescription drugs
Everything what you want to know about pills. Everything what you want to know about pills.
lipitor ilaci lipitor elea lipitor markings
neurontin and sciatica neurontin drug bank gabapentin glucosamine
can i take magnesium with pregabalin are a1 and lyrica still together 2022 what is the medicine pregabalin
dom valacyclovir valtrex japan valtrex autismweb
What side effects can this medication cause? Learn about the side effects, dosages, and interactions.
prescription drugs
Long-Term Effects. Commonly Used Drugs Charts.
synthroid tmax synthroid toxicity symptoms synthroid head tremors
Everything what you want to know about pills. Comprehensive side effect and adverse reaction information.
non prescription erection pills
Read information now. Top 100 Searched Drugs.
flagyl tiermedizin metronidazole components metronidazole derivatives
lipitor magyarul lipitor halveringstid lipitor b12
Get warning information here. Get here.
reputable canadian online pharmacy
п»їMedicament prescribing information. What side effects can this medication cause?
neurontin and vicoprofen gabapentin ilac lawsuits involving neurontin
how long does pregabalin take to kick in gabapentin and lyrica how long does lyrica stay in your system
valtrex lexapro valtrex duration valtrex caplets
Drug information. Read here.
https://canadianfast.com/# cat antibiotics without pet prescription
Read now. Best and news about drug.
hey there and thank you for your information – I have definitely picked
up something new from right here. I did however expertise a few technical points using this
web site, as I experienced to reload the web site a lot of times
previous to I could get it to load correctly. I had been wondering if your web hosting is OK?
Not that I am complaining, but sluggish loading instances times will often affect your placement in google and could
damage your high quality score if advertising and marketing with Adwords.
Well I am adding this RSS to my e-mail and could look out for much more of your respective intriguing content.
Make sure you update this again very soon.
I like the valuable info you provide in your articles. I’ll
bookmark your blog and check again here frequently.
I am quite sure I’ll learn plenty of new stuff right here!
Best of luck for the next!
Hola! I’ve been reading your site for some time now and finally got the courage to go ahead and give you a shout out from Kingwood Tx!
Just wanted to say keep up the good job!
I am in fact glad to read this weblog posts which contains plenty of valuable data,
thanks for providing these kinds of statistics.
Every weekend i used to visit this web page, for
the reason that i wish for enjoyment, for the reason that
this this website conations in fact pleasant funny stuff too.
Great blog you’ve got here.. It’s difficult to
find good quality writing like yours these days.
I seriously appreciate individuals like you! Take care!!
персонажи стар варс
Everything what you want to know about pills. Prescription Drug Information, Interactions & Side.
sildenafil 100 coupon
Read now. Get information now.
Do you mind if I quote a couple of your posts as long as
I provide credit and sources back to your blog?
My blog is in the very same niche as yours and my
visitors would really benefit from some of the information you provide here.
Please let me know if this alright with you. Thank you!
смотреть фильмы онлайн 2018 бесплатно в хорошем качестве ivi
Read information now. earch our drug database.
where to buy viagra
Read here. Prescription Drug Information, Interactions & Side.
Cautions. Read information now.
https://viagrapillsild.com/# government funded viagra
Cautions. Get warning information here.
essay online help essay review service online help with essay writing
Пацаны 4 сезон
Everything what you want to know about pills. Comprehensive side effect and adverse reaction information.
sildenafil 200mg for sale
earch our drug database. Read now.
paroxetine 12.5
best college essay editing service essay writers service video essay
scholarship essay whats a hook in an essay academic essay writer
word count essay short essay format common app essay examples
Drugs information sheet. Prescription Drug Information, Interactions & Side.
https://tadalafil1st.com/# cialis buy
Get here. Get information now.
4 paragraph essay mla format essay essay helper
why umich essay parts of an essay or letter for short profile essay
All trends of medicament. safe and effective drugs are available.
best price cialis 20mg
Top 100 Searched Drugs. Commonly Used Drugs Charts.
top essay writing services essay extender cover page for essay
real parchment paper for writing star writing paper free printable writing paper with borders
writing a seminar paper online research paper writer special writing paper
best online essay writing services essay help websites example of 200 word essay
legit essay writing services another word for essay how to structure an essay
generic propecia for sale
Read information now. Everything about medicine.
https://tadalafil1st.online/# what is in cialis ingredients
Drug information. Everything what you want to know about pills.
Get here. earch our drug database.
tadalafil soft gel
Get information now. Drug information.
how to buy a research paper buy college research papers writing a 10 page paper in one night
writing a position paper parchment paper for writing paper with writing on it
need help writing paper custom essay research paper parchment paper with writing
pay someone to write my research paper term papers for sale paper writing app
writing an introduction for a research paper writing a research paper in political science baglione pdf printable cursive writing paper
Everything about medicine. Learn about the side effects, dosages, and interactions.
https://tadalafil1st.online/# tadalafil 40 mg online india
Definitive journal of drugs and therapeutics. drug information and news for professionals and consumers.
Read now. Long-Term Effects.
https://tadalafil1st.com/# cialis original online
п»їMedicament prescribing information. Comprehensive side effect and adverse reaction information.
Get warning information here. Prescription Drug Information, Interactions & Side.
https://tadalafil1st.com/# paypal cialis payment
Long-Term Effects. Best and news about drug.
2181
acls mellon dissertation wiki conceptual framework dissertation dissertation planner
purchase research paper online term paper writing help writing services for college papers
literature review dissertation dissertation committee completing your qualitative dissertation
alphabet writing paper how to find someone to write my paper kindergarten writing paper with picture box
umi dissertation publishing how long is a dissertation masters dissertation
Метафора
writing a conclusion to a research paper custom term paper writing purchase custom research paper
Everything what you want to know about pills. Definitive journal of drugs and therapeutics.
amoxicillin 500 mg tablet price
earch our drug database. Get here.
dissertation team dissertation dГ©finition my dissertation is killing me
Stromectol 12 mg Stromectol over the counter: How do I know if my liver is repairing
dissertation binding dissertation template sample dissertation timeline
doctoral thesis vs dissertation dissertation topics in educational leadership dissertation statistics help
write my paper online paper writers online buy academic papers
п»їMedicament prescribing information. Get warning information here.
can we buy amoxcillin 500mg on ebay without prescription
Long-Term Effects. Read here.
what is the difference between a thesis and a dissertation dissertation prospectus sample do you italicize dissertation titles
help writing college research paper leprechaun writing paper how to writing paper
top ten essay writing services help writing grad school essay macbeth essay help
cat essay writer custom essay writing company custom essay writing canada
online essay help chat need help writing an essay online essay editing services
help with writing college essays help writing my college essay essay writer funny
nursing essay help professional essay editing service act essay help
online essay services best websites for essays buy cheap essays
Cool. I spent a long time looking for relevant content and found that your article gave me new ideas, which is very helpful for my research. I think my thesis can be completed more smoothly. Thank you.
fildena canada sildenafil 100mg uk fildena super active 100mg.
cheapest cialis available My heart goes out to those who didn t get their hair back
What happens if parasites are left untreated Hydroxychloroquine manufacturer?
cheap custom essay papers essay writing services singapore help with college essays
the best essay writer my custom essay best cheap essay
sildenafil amazon buy sildenafil over the counter viagra substitute walgreens
viagra canadian pharmacy ezzz sildenafil without prescription viagra for men
viagra naturel homme comment remplacer le viagra naturellement viagra online.
When should I worry about my liver azithromycin tablets – https://zithrominimax.com/
clomiphene citrate for women
plaquenil visual field what is hydroxychloroquine prescribed for plaquenil pill
cheap custom essay papers custom essays online essay writing service review
custom essays writing write my essay website admission essay editing service
Психолог онлайн
help writing grad school essay help writing a narrative essay english essay writing service
best paper writing site auto essay writer essay writing website reviews
help to write essay mba essay review service psychology essay writing services
How do you tell if a guy wants you to leave him alone cialis pill
writing a good thesis online thesis help how to pronounce thesis
rhb
three prong thesis components of a thesis statement thesis writing uk
graduation thesis thesis adviser hillary’s thesis
dissertation committee request email sample dissertation format sample how to cite dissertation apa
components of a thesis statement introduction and thesis statement example psychology thesis topics
hydroxychloroquine manufacturer hydroxychloroquine azithromycin zinc study hydroxychloroquine generic name
Can 1 baby have 2 fathers viagra for men near me
What would bring on an asthma attack albuterol over the counter equivalent
where can i buy cipro online
senior thesis topic thesis title definition of a thesis
buy silagra online
biaxin bronchitis
how to get amoxicillin over the counter
hydroxychloroquine brand name what is the generic name for plaquenil plaquenil hydroxychloroquine sulfate
no prescription needed canadian pharmacy
nolvadex medicine online
cost of phenergan tablets
antabuse tablets in india
tetracycline cost uk
tor market url dark web search engine
Can kidneys heal after quitting drinking combivent respimat
abilify generic 20 mg
Pills information leaflet. Cautions. is plaquenil an immunosuppressant Canada A-one trends of meds. On now.
strattera 40 mg pills
vermox united states
cafergot tablets price
ed meds: meds online without doctor prescription – ed meds online canada
hydroxychloroquine drugs what is hydroxychloroquine 200 mg plaquenil side effect
21 amoxicillin 500mg capsules
diflucan mexico
nexium prescription
prescription drugs online: buy prescription drugs online without – ed remedies that really work
inderal 60 mg
buy cytotec pills
price of prozac in india
tadacip 20 uk
erythromycin pills
online med pharmacy
tadacip 20 mg tablets
dapoxetine cream
cialis 10 mg cialis 10 mg http://cialis10fr.com/
generic zithromax 500mg use of azithromycin strep azithromycin
atenolol 25 mg tablet cost
tadalafil 60
erythromycin 500mg generic
ivermectin 80 mg
pharmacy express
generic avodart price
sexual dysfunction in men: cheap pet meds without vet prescription – ed natural treatment
dexamethasone 0.005
glucophage cheapest price
avodart uk prescription
dapoxetine tablets cost in india
Meds info sheet. Cautions. canadian pharmacy norco
ciprofloxacin 100mg price
40mg fluoxetine
buy nolvadex online usa
can you buy celexa online
rx robaxin
buy prozac online europe
Acheter viagra en ligne livraison 24h: Viagra homme prix en pharmacie sans ordonnance – Viagra 100 mg sans ordonnance
164 avana
benicar prescription
comprar viagra online en andorra: se puede comprar sildenafil sin receta – viagra entrega inmediata
best pharmacy
cleocin cream over the counter
SildГ©nafil 100 mg sans ordonnance: Viagra gГ©nГ©rique sans ordonnance en pharmacie – Acheter viagra en ligne livraison 24h
sildenafilo 50 mg comprar online: se puede comprar sildenafil sin receta – viagra para hombre venta libre
where can i buy cytotec in south africa
motilium price singapore
sildenafil vs cialis can you overdose on sildenafil sildenafil time to work
where to buy clonidine online
combining sildenafil and tadalafil bluechew tadalafil expiration date tadalafil 20 mg walmart
Viagra gГ©nГ©rique sans ordonnance en pharmacie: Viagra sans ordonnance 24h Amazon – Viagra gГ©nГ©rique sans ordonnance en pharmacie
cheap online pharmacy
order anafranil from canada
sildenafilo cinfa 100 mg precio farmacia: comprar viagra en espaГ±a envio urgente contrareembolso – viagra online gibraltar
how to get tadalafil out of your system cialis 20mg tadalafil daily dose of tadalafil for erectile dysfunction
tadalafil 20mg australia can you mix tadalafil and sildenafil low dose tadalafil
fluoxetine 60 mg cost
humana mail order pharmacy mills family pharmacy uva pharmacy
Acheter viagra en ligne livraison 24h: Viagra gГ©nГ©rique sans ordonnance en pharmacie – Viagra gГ©nГ©rique sans ordonnance en pharmacie
hydroxychloroquine & azithromycin plaquenil generic name hydroxychloroquine for sale in usa
furosemide spc furosemide 20 milligrams https://furosemide.directory
clonidine 0.5 mg
offshore pharmacy no prescription
viagra online cerca de zaragoza: Viagra online cerca de Madrid – Viagra online cerca de Madrid
Viagra sans ordonnance 24h Amazon: Viagra homme prix en pharmacie – Prix du Viagra en pharmacie en France
ampicillin brand name in india
tadalafil wiki how much tadalafil should i take raw tadalafil powder
buy accutane pills online
allopurinol 300 mg for sale without prescription
comprar viagra en espaГ±a: sildenafilo precio farmacia – comprar viagra en espaГ±a
how much is cozaar
hydroxychloroquine for lupus plaquenil tablets 200mg plaquenil dosage
propranolol tablets 20 mg
sildenafilo 100mg sin receta: comprar viagra sin gastos de envГo – sildenafilo cinfa 100 mg precio farmacia
Viagra 100mg prix: Viagra sans ordonnance 24h suisse – Le gГ©nГ©rique de Viagra
hydrochlorothiazide 50 mg price
venta de viagra a domicilio: viagra precio 2022 – viagra online cerca de bilbao
does tadalafil give you energy https://justtadafilix.com/ sublingual tadalafil
price of accutane
where to buy viagra without a prescription https://foxviagrixed.com/ buy viagra uk
generic zoloft 200 mg
elimite over the counter
sildenafil 100 no prescription https://ac3vigra.com/ female viagra online canada
cost of phenergan prescription
buy buspar online
buy elavil online cheap
I was looking for another article by chance and found your article slotsite I am writing on this topic, so I think it will help a lot. I leave my blog address below. Please visit once.
clomid canada over the counter
fildena 150
sildenafilo precio farmacia: comprar viagra en espaГ±a envio urgente – comprar viagra en espaГ±a envio urgente contrareembolso
price viagra generic https://leepvigras.com/ how do i get viagra without a prescription
cymbalta 60 mg cost canada
waterproof speaker
viagra sildenafil vs cialis sildenafil citrate side effects
finasteride 5mg over the counter
zofran medicine price
buy fildena 50 mg
resume for customer service manager cover letter writing job resume objective for customer service
comprar viagra sin gastos de envГo: farmacia gibraltar online viagra – sildenafilo 100mg farmacia
avodart medication generic
Can you be more specific about the content of your article? After reading it, I still have some doubts. Hope you can help me.
cialis dosage reddit https://crocilismen.com/ is cialis covered by medicare
strattera 60 mg
cialis in ireland https://hdcillis.com/ cost of cialis at costco
arimidex generic india
viagra generic uk https://ethvigrix.com/ cheap sildenafil canada
allopurinol over the counter
fildena 100 mg for sale
comprar viagra en espaГ±a amazon: comprar viagra online en andorra – sildenafilo 100mg precio farmacia
arimidex for sale australia
buy malegra online 100mg
tenormin anxiety
plaquenil manufacturers hydroxychloroquine 200mg where to buy hydroxychloroquine 200 mg
hydrochlorothiazide 25mg coupon
viagra online cerca de toledo: comprar viagra contrareembolso 48 horas – venta de viagra a domicilio
tamoxifen 20 mg tablet price in india
generic zestoretic
https://viasenzaricetta.com/# viagra online consegna rapida
buy generic levaquin online
miglior sito dove acquistare viagra viagra subito cerco viagra a buon prezzo
buy real viagra from canada https://vivigrix.com/ women viagra online no prescription
inderal best price
abilify 2mg tablet cost
accutane 120 mg
how long does sildenafil take to work roman sildenafil sildenafil dose for high blood pressure
cialis 5mg https://wwcillisa.com/ tadalafil from nootropic review
voltaren gel 60 mg
miglior sito per comprare viagra online viagra 50 mg prezzo in farmacia kamagra senza ricetta in farmacia
propecia usa buy
flomax price comparison
https://viasenzaricetta.com/# viagra ordine telefonico
viagra generico recensioni siti sicuri per comprare viagra online gel per erezione in farmacia
zovirax ointment cost
cheap cialis dapoxitine cheap online https://uhdcilise.com/ cialis stopped working
canadian pharmacy generic cialis https://hoscillia.com/ cialis tadalafil 10mg tablets
buy proscar 5mg proscar sale uk proscar 5 mg tablet cost
kamagra 100mg oral jelly suppliers
wellbutrin 75 mg tablets
atarax 50 mg price
medrol 4mg cost
buy atarax tablets uk
http://doxycyclinesale.pro/# doxycycline capsules 50mg 100mg
a prokaryotic cell r pro cars finasteride permanent side effects
citalopram 20 mg tab
diflucan 150 mg canada
happy family store pharmacy
amoxicillin canada prescription
tamoxifen price in india
where can i get diflucan online
how to get clomid prescription
generic zithromax 250mg
furosemide prescription medicine
Браво, эта замечательная фраза придется как раз кстати
kapelki-firefit.ru
И что бы мы делали без вашей отличной идеи
kapelki-firefit.ru
What can I drink to have flat tummy Cenforce 100 canada?
What a man wants in a woman he wants to marry order generic Cenforce?
What is the best love word buy vidalista-20?
brillx casino официальный мобильная версия
бриллкс
Но если вы готовы испытать настоящий азарт и почувствовать вкус победы, то регистрация на Brillx Казино откроет вам доступ к захватывающему миру игр на деньги. Сделайте свои ставки, и каждый спин превратится в захватывающее приключение, где удача и мастерство сплетаются в уникальную симфонию успеха!Брилкс казино предоставляет выгодные бонусы и акции для всех игроков. У нас вы найдете не только классические слоты, но и современные игровые разработки с прогрессивными джекпотами. Так что, возможно, именно здесь вас ждет величайший выигрыш, который изменит вашу жизнь навсегда!
A lowrisk subset of patients in whom RT may be omitted without compromising local control remains to be defined how to buy priligy in usa
Is erectile dysfunction a sign of underlying heart disease order fildena generic?
What will happen when you meet your soulmate fildena buy?
Treatments for ED may include oral medications like Viagra, Cialis, or Levitra, which enhance blood flow to the penis. priligy for sale
the feeling of soft sand between your toes is soothing and grounding instruction where google translat
Researchers are investigating the effects of chronic hormonal imbalances, such as elevated prolactin levels, on erectile function. Addressing hormonal abnormalities through medication or other interventions can help restore normal sexual function. how to buy priligy
Рекомендую Вам поискать в google.com
https://kapelki-firefit.ru/
это хорошо
kapelki-firefit.ru
dog prednisone dosage cancer can i take antacid with prednisone does prednisone help sinus infection
viagra 100mg tablet buy online viagra tablet purchase online buy sildenafil 20 mg without prescription
Can antibiotics be used for cellulitis buy hydroxychloroquine 200 mg
sildenafil 100mg from india sildenafil generic for sale viagra substitutes
where to buy viagra online uk generic viagra online united states viagra pills for sale uk
Can antibiotics be used to treat histoplasmosis plaquenil 200mg price in ksa
viagra for women uk viagra soft tabs sildenafil 90 mg
pharmacy support group viagra pharmacy assistant course online online pharmacy products
buy soma mexican pharmacy provera online pharmacy cancun pharmacy vicodin
health express pharmacy artane japan pharmacy online target pharmacy clonazepam
cialis drug order original cialis purchase cialis online canadian
cialis ed cialis comparison when is generic cialis available
liquid cialis cialis delivered in 24 hours canadian online pharmacy cialis
tadalafil tablets ip cialis dosage reddit cialis canada
I am currently writing a paper and a bug appeared in the paper. I found what I wanted from your article. Thank you very much. Your article gave me a lot of inspiration. But hope you can explain your point in more detail because I have some questions, thank you. 20bet
Including disrespect, dishonesty, controlling behaviors, or a absence of assistance.
In a healthy partnership, everything just kind of works. Certain, you may disagree from period to time or arrive upon additional bumps in the street, but you usually jointly make decisions, discuss any issues that arise openly, and honestly take pleasure in each other’t company.
1321
http://gracetea.com/catalogsearch/result/index/?q=Buy%20Dapoxetine%20hcl%2030mg%20-%20www.RxLara.com%20-%20Priligy%2060%20%20tablet.%20For%20Premature%20Ejaculation%20OTC%20-%20www.rxlara.com priligy 60 mg price. These herbs have been used for centuries to improve sexual function and have been shown to be effective in treating premature ejaculation..
Inhaler Additives: The Pros and Cons of Using Flavoring Agents ventolin inhaler 100mcg.
What makes asthma worse at night albuterol sulfate hfa.
What keeps your prostate healthy Cenforce 150 good or bad.
%%
Take a look at my blog … ramblermails.com
How can I save my sperm Cenforce 100mg tablet.
How can I make my husband feel a man again order generic Cenforce.
What are the basics of quality order Cenforce 100mg pills.
They play a role in pain management for individuals with chronic migraines and headaches https://filmfreeway.com/Ivermectin-UnravelingthePotentialofaProvenAntibiotic stromectol 3mg information.
Healthcare providers should actively involve patients and caregivers in medication reconciliation during care transitions https://radiopaedia.org/users/stromectol-ivermectin stromectol cvs.
This design is wicked! You obviously know how to keep a
reader amused. Between your wit and your videos, I was almost moved to start my own blog (well,
almost…HaHa!) Wonderful job. I really enjoyed
what you had to say, and more than that, how you presented it.
Too cool!
филиппин
Ebony big butts have lots of various names- fanny, rear end, buns, and booty.https://www.studentitop.it/grammatica/come-fare-un-testo-argomentativo-regole-ed-esempi/ Fanny is a phrase that’s more mainstream and less intimate, so anyone can use it. Bottom is usually generally utilized in a jokey method. Buns sound even more sexy, and booty can be noticed as naughty. In Traditional western culture, the dimension of a woman’s rear end, boobs, and waist dimension usually determines how attractive she is. White females tend to choose larger boobs and a regular dimension butt, as long as it’s not really toned. But ebony females are usually proud of their large, juicy butts and like to show them off. It’s something they make use of to their benefit, and porno fans all over the global entire world understand this. Look at J Just. Lo – she may become a excellent vocalist and dancer, but it’s her well-known behind that put her in the spotlight. It’s even rumored to be covered!
High-speed sachet filling machines are used for packaging powdered or granular medications in convenient single-dose sachets stromectol 12mg online
Ebony large butts have lots of different names- fanny, booty, buns, and booty.https://www.petra-fabinger.de/2018/03/04/kunden-offerten-im-maerz/ Fanny is a phrase that’s more mainstream and much less sexual, so anyone can make use of it. Booty is used in a jokey way usually. Buns audio even more sexy, and booty can be seen as kinky. In Western tradition, the dimension of a woman’s bottom, boobs, and waist size determines how desirable she is usually. White women are likely to prefer bigger boobs and a regular size rear end, as lengthy as it’s not smooth. But ebony ladies are proud of their huge, juicy love and butts to show them off. It’s something they make use of to their benefit, and porno followers all over the globe understand this. Just look at J. Lo – she may end up being a excellent singer and dancer, but it’s her popular booty that place her in the spotlight. It’s actually rumored to become insured!
Hey there, You’ve done a fantastic job. I will certainly digg it and personally recommend to my friends.
I’m sure they’ll be benefited from this site.
Medications for allergies provide relief from symptoms like sneezing, itching, and congestion cheap Cenforce 50mg.
Students should learn about the risks associated with medication shortages and the role of healthcare professionals in managing them Cenforce 200 sildenafil citrate.
Семейный психолог в москве цены
Сколько стоит сеанс психолога
I really like reading through a post that can make men and women think. Also, thank you for allowing me to comment!
búp bê sexy
buy kamagra 100mg oral jelly uk https://community.alteryx.com/t5/user/viewprofilepage/user-id/524741/ symbicort coupons printable
You can email the site owner to let them know you were blocked. Please include what you were doing when this page came up and the Cloudflare Ray ID found at the bottom of this page. Fav+ 3081 ◈ The best casino. Come and enjoy a variety of thrilling casino games in Cash Bay Casino.. Cash Bay Casino is suitable for ages 17+. It’s filesize is 146.67 MB. Seamless sync across devices* There are several ways for players to win big prizes in Casino Frenzy-Slot,Poker,Bingo. From world-famous slot games such as Aladdin Slots, Ganesh Slots, and Fruit Slots, you’ll enjoy diverse jackpots waiting to be won. The app also features a classic fishing game, and popular card games such as poker, baccarat, dragon tiger, and Texas Hold’em, where you can also compete with other players.
https://joycesulysses.com/community/profile/ktowlichwohnsmo
LuckyLand Slots gives players multiple opportunities to obtain FREE Sweeps Coins. Here’s a simple step-by-step process for using this bonus at LuckyLand Slots casino: This is easily one of the most user-friendly casino bonuses we’ve come across. This is because you can use the coins on all of the slot games featured at Luckyland with no restrictions. Plus there are no limits on how many coins you are allowed to wager at a time on these slots. Welcome to the ultimate guide on how to win coins at LuckyLand Slots! Our aim is to equip you with practical strategies, insights and tips that will improve your gameplay and increase your overall time playing by helping you bolster the number of coins you can play with. Like many social casinos, LuckyLand Slots has a strong social media presence. Whether you’re just joining the fun over at LuckyLand Slots or you’ve been a longtime member, we recommend that you link your Facebook account to your LuckyLand Slots player account. Not only is it a great way to stay current on all of the latest LuckyLand promotions, but you will also be notified about new games and tournaments as they become available.
How are medications tailored to meet the needs of specific patient populations?
How do I measure the correct dosage for liquid medications without a syringe?
Thanks so much for giving everyone a very wonderful possiblity to read in detail from this website. It is often so good and as well , jam-packed with amusement for me and my office mates to visit your site at the least thrice per week to read the newest items you have got. Of course, I’m certainly fascinated considering the splendid tricks served by you. Certain two ideas in this article are undoubtedly the most suitable we have all had.
How do people act when they have syphilis plaquenil cost assistance?
kitchen aids have a variety of different appliances that can help you cook your food easier`
Thanks for creating this. I really feel as though I know so much more about the topic than I did before. You should continue this, Im sure most people would agree youve got a gift.
Thank you for your site post. Jones and I are actually saving to get a new publication on this issue and your post has made all of us to save our own money. Your notions really clarified all our questions. In fact, more than what we had thought of prior to when we stumbled on your wonderful blog. We no longer nurture doubts as well as a troubled mind because you have actually attended to our own needs in this post. Thanks
???? ?????? ?????? ??? ??????? ?????????? ?? ????? ????? ????? ?? ????? ??????
What should I know before giving antibiotics retail cost of plaquenil?
Hiya. Very nice blog!! Guy .. Excellent .. Amazing .. I’ll bookmark your blog and take the feeds additionally…I am glad to find so much helpful information right here in the post. Thank you for sharing.
Can antibiotics be used for impetigo in children buy ivermectin?
Can antibiotics cause sun sensitivity buy stromectol 3mg online?
dapoxetine
Thank you, I have recently been looking for info about this subject for ages and yours is the best I’ve discovered till now. But, what about the conclusion? Are you sure about the source?
I do not understand exactly how I ran across your blog because I had been researching information on Real Estate inWinter Springs, FL, but anyway, I have enjoyed reading it, keep it up!
mobile devices are always great because they always come in a handy package’
What is the smallest age gap between siblings kamagra for woman
Locate which will article writing is not actually valued at the time period, you might recognise that discovering them free of charge is really a lot a great deal more an individual’s idea! Precisely why craft content articles together with throw away your time and energy when you are able become terrific content absolutely free! A lot of people discover utilizing just a dash of effort paid out diving the web page and looking at what’s going on they is able to leave and reveal a whole lot of function done with the particular means available.
The the next time I read a weblog, Hopefully that it doesnt disappoint me around this blog. I mean, It was my replacement for read, but When i thought youd have some thing fascinating to mention. All I hear can be a couple of whining about something you could fix if you werent too busy in search of attention.
I am glad to be a visitor of this gross weblog, appreciate it for this rare info!
persuasive essay community service https://mixbookmark.com/story1387719/not-known-factual-statements-about-url coconut tree essay in english
I like what you guys are up too. Such smart work and reporting! Carry on the superb works guys I?ve incorporated you guys to my blogroll. I think it will improve the value of my web site
Along with every little thing that appears to be building within this particular area, many of your opinions are generally fairly radical. Nevertheless, I am sorry, but I do not subscribe to your whole strategy, all be it radical none the less. It looks to everyone that your remarks are generally not completely rationalized and in simple fact you are your self not even entirely convinced of the argument. In any case I did enjoy looking at it.
Great paintings! This is the type of information that are meant to be shared across the net. Shame on the seek for now not positioning this submit upper! Come on over and consult with my site . Thanks =)
I am glad for writing to let you be aware of of the magnificent discovery my princess experienced using the blog. She came to find plenty of issues, not to mention what it’s like to possess a great teaching character to get many others just know a number of advanced things. You really did more than visitors’ expectations. Thank you for giving such insightful, healthy, revealing as well as fun guidance on this topic to Lizeth.
I discovered your blog post web site on the internet and appearance some of your early posts. Always maintain within the good operate. I just extra the Feed to my MSN News Reader. Looking for toward reading much more on your part down the line!…
The great news on News is very much imptortant to us.
Just like the old saying goes, within the pro’s head there are few options, however , for a person with the beginner’s brain, the world is open up.
Does a one day job count as “Work Experience” under the common application?
The next time I read a blog, I hope which it doesnt disappoint me up to this blog. I mean, I know it was my option to read, but I personally thought youd have something fascinating to state. All I hear is often a handful of whining about something that you could fix if you werent too busy interested in attention.
“I’m just writing to make you understand what a helpful experience my princess enjoyed reading through your web page. She learned a lot of pieces, including what it is like to have an ideal teaching heart to get other individuals with no trouble learn selected impossible subject areas. You truly surpassed readers’ desires. Many thanks for churning out such invaluable, healthy, informative and fun tips on the topic to Julie.”
I have been checking out some of your articles and i must say pretty good stuff. I will definitely bookmark your blog.
Whoa. That was a great article. Please keep writing because I love your style.
essay on christmas day for kids https://https-payformyessayser-co47024.spintheblog.com/23834906/everything-about-pay-people-to-write-essay essays on the problem of evil
Advancements in Orthopedic Surgery – Restoring Mobility how to get Cheap Clomid
Hi there, thanks for the awesome posting. I’m having troubles subscribing to your blogs feed. Thought I’d let you know
whoah this blog is great i love reading your articles. Keep up the good work! You know, lots of people are looking around for this info, you could aid them greatly.
Keep websiteing stuff like this I actually am fond of it
I really appreciate this particular blog and exactly how its laid out, thankyou I’ve saved.
cheers for such a great blog. Where else could anyone get that kind of info written in such a perfect way? I have a presentation that I am presently working on, and I have been on the look out for such info.
Good – I should definitely say I’m impressed with your web site. I had no trouble navigating through all the tabs and related information. The site ended up being truly easy to access. Nice job.
I am frequently to blogging i actually appreciate your articles. This article has truly peaks my interest. I am going to bookmark your web blog and keep checking for first time information.
I have been exploring for a little bit for any high-quality articles or blog posts on this sort of area . Exploring in Yahoo I at last stumbled upon this website. Reading this information So i am happy to convey that I’ve an incredibly good uncanny feeling I discovered exactly what I needed. I most certainly will make certain to do not forget this web site and give it a glance regularly.
I think this is among the most significant information for me. And i am glad reading your article. But want to remark on some general things, The web site style is perfect, the articles is really nice : D. Good job, cheers
prednisone 2 mg daily: where to buy prednisone in australia – prednisone 50mg cost
http://ciprofloxacin.life/# where can i buy cipro online
can i purchase clomid without rx: can you get cheap clomid prices – can i purchase clomid
where can i buy cheap clomid without prescription where can i buy generic clomid without insurance – get generic clomid without prescription
http://amoxil.icu/# amoxicillin over the counter in canada
http://amoxil.icu/# can i buy amoxicillin online
buy doxycycline monohydrate: buy doxycycline online uk – purchase doxycycline online
can i order lisinopril over the counter zestoretic 10 12.5 mg lisinopril tabs 20mg
doxycycline 100mg tablets: doxycycline tetracycline – doxycycline vibramycin
http://doxycyclinebestprice.pro/# doxycycline hyc 100mg
Somebody necessarily lend a hand to make significantly posts I
would state. This is the very first time I frequented your website page
and thus far? I amazed with the research you
made to create this particular submit amazing.
Excellent activity!
zithromax generic price: zithromax for sale cheap – how to get zithromax
http://cytotec.icu/# buy cytotec over the counter
lisinopril brand name cost: lisinopril 5mg pill – lisinopril 20 mg coupon
Cytotec 200mcg price: order cytotec online – cytotec online
http://lisinoprilbestprice.store/# 208 lisinopril
zithromax 500 mg lowest price pharmacy online: zithromax online no prescription – buy cheap generic zithromax
zithromax pill zithromax z-pak zithromax drug
http://zithromaxbestprice.icu/# buy zithromax online with mastercard
tamoxifen cost: hysterectomy after breast cancer tamoxifen – nolvadex only pct
doxycycline 50 mg: buy doxycycline 100mg – doxycycline 500mg
http://doxycyclinebestprice.pro/# doxycycline hyc 100mg
doxycycline 50 mg: doxycycline 100mg capsules – order doxycycline online
clomid nolvadex: tamoxifen for men – does tamoxifen cause weight loss
purchase cytotec cytotec online buy cytotec over the counter
http://cytotec.icu/# Misoprostol 200 mg buy online
order doxycycline online: doxycycline 200 mg – doxycycline 50mg
http://cytotec.icu/# purchase cytotec
buy misoprostol over the counter: buy cytotec in usa – buy cytotec over the counter
tamoxifen therapy: aromatase inhibitors tamoxifen – tamoxifen benefits
reputable canadian pharmacy: canadian discount pharmacy – canadian pharmacy india canadapharm.life
https://indiapharm.llc/# best online pharmacy india indiapharm.llc
mexico drug stores pharmacies: mexican pharmacy – mexico pharmacy mexicopharm.com
mexican rx online: Best pharmacy in Mexico – mexican rx online mexicopharm.com
best online pharmacies in mexico: Mexico pharmacy online – mexico pharmacies prescription drugs mexicopharm.com
buying drugs from canada Pharmacies in Canada that ship to the US northern pharmacy canada canadapharm.life
https://canadapharm.life/# canadian world pharmacy canadapharm.life
indianpharmacy com: India pharmacy of the world – online pharmacy india indiapharm.llc
https://canadapharm.life/# canadian drug pharmacy canadapharm.life
best mail order pharmacy canada: Canadian online pharmacy – drugs from canada canadapharm.life
https://mexicopharm.com/# mexican pharmaceuticals online mexicopharm.com
canadian pharmacy service: precription drugs from canada – onlinecanadianpharmacy canadapharm.life
world pharmacy india: Online India pharmacy – buy medicines online in india indiapharm.llc
pharmacy com canada: Canada Drugs Direct – canadian drug pharmacy canadapharm.life
mexican border pharmacies shipping to usa Mexico pharmacy online mexican drugstore online mexicopharm.com
http://indiapharm.llc/# Online medicine order indiapharm.llc
How are bionic eyes changing the lives of individuals with visual impairments viagra over the counter united states
legal canadian pharmacy online: best canadian online pharmacy – canadian pharmacy oxycodone canadapharm.life
https://canadapharm.life/# best canadian pharmacy to buy from canadapharm.life
mexican drugstore online: mexican pharmacy – best online pharmacies in mexico mexicopharm.com
indian pharmacies safe: indian pharmacy to usa – indian pharmacy indiapharm.llc
medicine in mexico pharmacies: Mexico pharmacy online – mexico drug stores pharmacies mexicopharm.com
online shopping pharmacy india: Online India pharmacy – buy medicines online in india indiapharm.llc
http://indiapharm.llc/# online pharmacy india indiapharm.llc
canada drug pharmacy: Pharmacies in Canada that ship to the US – canada cloud pharmacy canadapharm.life
online shopping pharmacy india Medicines from India to USA online online pharmacy india indiapharm.llc
pharmacy tampa oxycodone
Kamagra 100mg: buy kamagra – super kamagra
https://edpillsdelivery.pro/# ed treatment drugs
sildenafil buy online without a prescription: Sildenafil price – sildenafil tablet in india
ed drugs: ed pills online – herbal ed treatment
Buy Vardenafil online Levitra best price buy Levitra over the counter
http://kamagradelivery.pro/# п»їkamagra
Levitra 10 mg buy online: Vardenafil online prescription – Levitra online pharmacy
https://levitradelivery.pro/# Cheap Levitra online
where to buy tadalafil in usa: Buy tadalafil online – tadalafil 10 mg canadian pharmacy
sildenafil 100 mg prescription: Cheapest Sildenafil online – sildenafil generic drug cost
https://tadalafildelivery.pro/# tadalafil 20mg price in india
online tadalafil prescription: cheap tadalafil canada – generic tadalafil for sale
what are ed drugs cheapest ed pills herbal ed treatment
Kamagra 100mg price: kamagra oral jelly – super kamagra
https://levitradelivery.pro/# Buy generic Levitra online
https://kamagradelivery.pro/# kamagra
ed drugs compared: erection pills over the counter – best ed treatment pills
http://levitradelivery.pro/# Levitra online pharmacy
sildenafil pfizer: Sildenafil price – buy sildenafil with visa
Vardenafil price: Buy generic Levitra online – Levitra 20 mg for sale
http://clomid.auction/# how to get generic clomid no prescription
paxlovid price paxlovid price without insurance paxlovid pill
https://stromectol.guru/# ivermectin 4000
stromectol tablets uk: ivermectin 6 – ivermectin purchase
http://clomid.auction/# can i get generic clomid pill
п»їpaxlovid paxlovid price without insurance paxlovid covid
http://clomid.auction/# buying cheap clomid without dr prescription
ivermectin 0.5: ivermectin for sale – ivermectin tablet price
http://paxlovid.guru/# paxlovid for sale
http://stromectol.guru/# ivermectin 20 mg
whoah this weblog is great i really like studying your posts. Keep up the good work! You recognize, lots of individuals are hunting around for this information, you could aid them greatly.
buy paxlovid online Paxlovid over the counter Paxlovid buy online
paxlovid price: Buy Paxlovid privately – paxlovid cost without insurance
https://amoxil.guru/# amoxicillin 500 mg without a prescription
https://stromectol.guru/# what is minocycline 50 mg used for
https://clomid.auction/# can i order generic clomid without rx
zithromax z-pak price without insurance: cheapest azithromycin – zithromax antibiotic
dapoxetine 60 mg sildenafil 100mg brands Continuous manufacturing systems have become more prevalent, improving efficiency and reducing waste.
cost of propecia prices Buy Finasteride 5mg cost generic propecia no prescription
http://azithromycin.store/# how to get zithromax over the counter
order propecia without rx: Buy finasteride 1mg – order cheap propecia prices
lisinopril 80 mg: High Blood Pressure – zestril coupon
https://azithromycin.store/# generic zithromax 500mg
lisinopril australia: where to buy lisinopril online – lisinopril 5
http://finasteride.men/# cost of propecia no prescription
zithromax drug zithromax best price zithromax for sale cheap
http://finasteride.men/# get cheap propecia no prescription
zithromax: zithromax best price – zithromax buy online
https://finasteride.men/# cost generic propecia prices
cytotec online: Misoprostol best price in pharmacy – Abortion pills online
zithromax generic price: buy zithromax z-pak online – generic zithromax over the counter
https://lisinopril.fun/# lisinopril 40mg
lasix online: Over The Counter Lasix – lasix side effects
cheap propecia pill Buy Finasteride 5mg get generic propecia pills
zithromax for sale online: buy zithromax z-pak online – generic zithromax medicine
https://azithromycin.store/# zithromax online no prescription
cost propecia prices: buy propecia – cost of propecia online
buying propecia without prescription: Buy Finasteride 5mg – cheap propecia online
http://azithromycin.store/# where can i purchase zithromax online
https://azithromycin.store/# buy zithromax without prescription online
lisinopril 60 mg: buy lisinopril online – order lisinopril
http://finasteride.men/# cost of cheap propecia price
zithromax 500 mg lowest price pharmacy online: Azithromycin 250 buy online – zithromax 250 mg australia
order lisinopril online us: over the counter lisinopril – lisinopril medication generic
generic propecia without dr prescription: Buy Finasteride 5mg – buying generic propecia pills
purchase cytotec buy misoprostol order cytotec online
https://lisinopril.fun/# lisinopril 20 mg canada
https://lisinopril.fun/# lisinopril 2.5 mg
get generic propecia without a prescription: Cheapest finasteride online – get propecia price
lasix 100 mg tablet: Buy Lasix No Prescription – lasix generic name
azithromycin zithromax Azithromycin 250 buy online zithromax 500mg price in india
how to get zithromax online: zithromax without prescription – zithromax 500mg price in india
lisinopril coupon: High Blood Pressure – prinivil brand name
http://sildenafilitalia.men/# viagra online spedizione gratuita
migliori farmacie online 2023: cialis generico consegna 48 ore – comprare farmaci online con ricetta
https://avanafilitalia.online/# acquisto farmaci con ricetta
comprare farmaci online con ricetta Dove acquistare Cialis online sicuro comprare farmaci online con ricetta
farmacia online: farmacia online migliore – farmacia online miglior prezzo
п»їfarmacia online migliore: farmacia online migliore – comprare farmaci online con ricetta
http://kamagraitalia.shop/# farmacie online sicure
http://farmaciaitalia.store/# acquisto farmaci con ricetta
п»їfarmacia online migliore: kamagra – п»їfarmacia online migliore
http://farmaciaitalia.store/# farmacia online più conveniente
acquisto farmaci con ricetta: kamagra gel – migliori farmacie online 2023
http://farmaciaitalia.store/# farmacia online senza ricetta
http://kamagraitalia.shop/# top farmacia online
farmacie online affidabili: farmacia online spedizione gratuita – farmacie on line spedizione gratuita
acquisto farmaci con ricetta: Tadalafil generico – п»їfarmacia online migliore
farmacie online autorizzate elenco: avanafil generico – farmacia online senza ricetta
http://kamagraitalia.shop/# farmacie online affidabili
comprare farmaci online con ricetta kamagra farmacie on line spedizione gratuita
farmacia online senza ricetta: top farmacia online – migliori farmacie online 2023
http://avanafilitalia.online/# farmacia online piГ№ conveniente
https://canadapharm.shop/# canadian pharmacies comparison
canadian pharmacy uk delivery cheap canadian pharmacy online adderall canadian pharmacy
best india pharmacy: india pharmacy – top 10 pharmacies in india
canada pharmacy reviews: canadian discount pharmacy – canadian pharmacy no scripts
https://mexicanpharm.store/# buying prescription drugs in mexico online
mexico drug stores pharmacies: pharmacies in mexico that ship to usa – mexican pharmacy
canadian online drugstore: canadian pharmacy meds reviews – canada pharmacy online
http://canadapharm.shop/# canadian pharmacy online
pharmacies in mexico that ship to usa best online pharmacies in mexico pharmacies in mexico that ship to usa
indian pharmacies safe: cheapest online pharmacy india – best india pharmacy
http://canadapharm.shop/# vipps canadian pharmacy
canadian online drugs: canadian pharmacy – canada ed drugs
indianpharmacy com: top 10 pharmacies in india – world pharmacy india
http://canadapharm.shop/# canadian pharmacy antibiotics
mexican rx online: mexican border pharmacies shipping to usa – buying prescription drugs in mexico online
cheapest online pharmacy india: canadian pharmacy india – india pharmacy
https://indiapharm.life/# п»їlegitimate online pharmacies india
buy canadian drugs: pharmacy rx world canada – trustworthy canadian pharmacy
mexico pharmacies prescription drugs: buying from online mexican pharmacy – purple pharmacy mexico price list
reliable canadian pharmacy canadian pharmacy 24h com safe canadian pharmacy prices
Online medicine home delivery: top 10 online pharmacy in india – best india pharmacy
https://canadapharm.shop/# canadianpharmacymeds com
indian pharmacy paypal: india pharmacy mail order – indian pharmacy online
canadian online pharmacy: canadian pharmacies online – best online canadian pharmacy
http://mexicanpharm.store/# best online pharmacies in mexico
indian pharmacy: indian pharmacy online – indian pharmacy online
http://mexicanpharm.store/# buying prescription drugs in mexico online
mexican mail order pharmacies: pharmacies in mexico that ship to usa – mexico drug stores pharmacies
mexican online pharmacies prescription drugs mexican border pharmacies shipping to usa mexican border pharmacies shipping to usa
mexican mail order pharmacies: mexico drug stores pharmacies – mexico drug stores pharmacies
https://mexicanpharm.store/# mexican pharmacy
canadian pharmacies compare: 77 canadian pharmacy – buying drugs from canada
http://indiapharm.life/# indian pharmacies safe
best rated canadian pharmacy: best rated canadian pharmacy – canadian pharmacies comparison
http://indiapharm.life/# india pharmacy mail order
http://prednisonepharm.store/# online order prednisone
A pharmacy that truly understands international needs http://prednisonepharm.store/# purchase prednisone 10mg
prednisone ordering online: prednisone 20mg nz – prednisone no rx
https://cytotec.directory/# buy cytotec pills
Their patient education resources are top-tier https://zithromaxpharm.online/# generic zithromax online paypal
https://prednisonepharm.store/# prednisone 12 mg
tamoxifen alternatives how to prevent hair loss while on tamoxifen where to buy nolvadex
https://zithromaxpharm.online/# buy zithromax online australia
zithromax without prescription: buy azithromycin zithromax – buy zithromax 500mg online
They simplify the complexities of international prescriptions http://prednisonepharm.store/# prednisone pack
http://cytotec.directory/# buy cytotec online
Their medication synchronization service is fantastic https://cytotec.directory/# Abortion pills online
http://nolvadex.pro/# tamoxifen alternatives
http://zithromaxpharm.online/# zithromax generic cost
get generic clomid without rx cost generic clomid for sale can i get clomid pills
Their commitment to global patient welfare is commendable http://cytotec.directory/# buy cytotec
https://zithromaxpharm.online/# buy generic zithromax no prescription
They provide valuable advice on international drug interactions https://prednisonepharm.store/# prednisone 5 mg tablet price
http://prednisonepharm.store/# generic over the counter prednisone
The go-to place for all my healthcare needs https://cytotec.directory/# buy cytotec online
http://zithromaxpharm.online/# zithromax purchase online
buy zithromax without prescription online buy generic zithromax no prescription zithromax capsules price
real canadian pharmacy: top 10 online pharmacies – northeast discount pharmacy
ed drugs ed medications list ed meds online
my canadian pharmacy online https://reputablepharmacies.online/# cheap prescriptions
internet pharmacies
http://reputablepharmacies.online/# best online pharmacy no prescription
real viagra without a doctor prescription usa: viagra without a doctor prescription – non prescription ed drugs
https://reputablepharmacies.online/# accredited canadian pharmacies
online pharmacies canadian usa pharmacy online verified canadian pharmacies
http://edwithoutdoctorprescription.store/# prescription drugs online
medications without prescription: prescription drugs online without doctor – list of canadian pharmacies
buy prescription drugs without doctor prescription drugs without doctor approval viagra without doctor prescription
prescription drugs online without: mexican pharmacy without prescription – meds online without doctor prescription
trustworthy canadian pharmacy https://edwithoutdoctorprescription.store/# ed meds online without doctor prescription
canadian neighbor pharmacy legit
https://edpills.bid/# treatment for ed
mexican pharmacy without prescription best ed pills non prescription buy prescription drugs
best over the counter ed pills: ed treatment review – new ed pills
largest canadian pharmacy canadian discount pharmacy canada drugs without prescription
http://edwithoutdoctorprescription.store/# ed meds online without doctor prescription
http://reputablepharmacies.online/# trustworthy canadian pharmacy
non prescription erection pills: best medication for ed – pills for ed
non prescription ed drugs real viagra without a doctor prescription usa buy prescription drugs without doctor
best male ed pills: ed medications online – men’s ed pills
online pharmacy no prescriptions: online pharmacies without an rx – online pharmacy usa
viagra without a doctor prescription walmart: best ed pills non prescription – non prescription ed drugs
http://edwithoutdoctorprescription.store/# prescription drugs without doctor approval
erection pills viagra online online ed pills online ed pills
medication for ed dysfunction: cheap ed pills – ed pills
ed drugs list top rated ed pills best ed medication
http://reputablepharmacies.online/# top 10 online pharmacies
https://reputablepharmacies.online/# online meds no rx reliable
purple pharmacy mexico price list: Medicines Mexico – mexican mail order pharmacies mexicanpharmacy.win
canadian pharmacy ltd canadian pharmacy checker canadian pharmacy 365 canadianpharmacy.pro
https://canadianpharmacy.pro/# canadianpharmacyworld canadianpharmacy.pro
https://mexicanpharmacy.win/# mexico drug stores pharmacies mexicanpharmacy.win
Online medicine home delivery: Best Indian pharmacy – india pharmacy indianpharmacy.shop
Ponto IPTV a melhor programacao de canais IPTV do Brasil, filmes, series, futebol
My website: порно с преподом русское
top online pharmacy india reputable indian pharmacies indian pharmacy indianpharmacy.shop
http://mexicanpharmacy.win/# medication from mexico pharmacy mexicanpharmacy.win
canada drug store
https://mexicanpharmacy.win/# mexican online pharmacies prescription drugs mexicanpharmacy.win
canadian pharmacy prices: ed meds online canada – canada drug pharmacy canadianpharmacy.pro
legitimate canadian pharmacy Cheapest drug prices Canada adderall canadian pharmacy canadianpharmacy.pro
ventolin inhaler online – How does the UK regulate the use of medicines during pregnancy and breastfeeding?
canadian online pharmacy Canada Pharmacy canadian mail order pharmacy canadianpharmacy.pro
http://mexicanpharmacy.win/# mexican rx online mexicanpharmacy.win
http://mexicanpharmacy.win/# mexican border pharmacies shipping to usa mexicanpharmacy.win
canadian online pharmacy Canada Pharmacy canadian pharmacies compare canadianpharmacy.pro
http://canadianpharmacy.pro/# pharmacy in canada canadianpharmacy.pro
https://canadianpharmacy.pro/# safe online pharmacies in canada canadianpharmacy.pro
aarp approved canadian online pharmacies
https://mexicanpharmacy.win/# mexican pharmacy mexicanpharmacy.win
https://mexicanpharmacy.win/# mexico pharmacy mexicanpharmacy.win
india online pharmacy
pharmacies in mexico that ship to usa mexico drug stores pharmacies mexico pharmacy mexicanpharmacy.win
https://canadianpharmacy.pro/# certified canadian pharmacy canadianpharmacy.pro
https://indianpharmacy.shop/# indian pharmacies safe indianpharmacy.shop
indian pharmacies safe
mexico drug stores pharmacies mexican pharmacy online п»їbest mexican online pharmacies mexicanpharmacy.win
https://indianpharmacy.shop/# reputable indian online pharmacy indianpharmacy.shop
top 10 pharmacies in india
https://indianpharmacy.shop/# indianpharmacy com indianpharmacy.shop
canadian pharmacy india pharmacy canadian superstore cross border pharmacy canada canadianpharmacy.pro
https://mexicanpharmacy.win/# medicine in mexico pharmacies mexicanpharmacy.win
http://mexicanpharmacy.win/# medicine in mexico pharmacies mexicanpharmacy.win
п»їlegitimate online pharmacies india
http://indianpharmacy.shop/# buy medicines online in india indianpharmacy.shop
mexican pharmacy mexican pharmacy online reputable mexican pharmacies online mexicanpharmacy.win
https://indianpharmacy.shop/# top 10 pharmacies in india indianpharmacy.shop
best india pharmacy
https://canadianpharmacy.pro/# legit canadian online pharmacy canadianpharmacy.pro
reputable indian online pharmacy Cheapest online pharmacy Online medicine home delivery indianpharmacy.shop
http://indianpharmacy.shop/# cheapest online pharmacy india indianpharmacy.shop
best online pharmacy without prescriptions
https://canadianpharmacy.pro/# canadian pharmacy online reviews canadianpharmacy.pro
canadian pharmacy online ship to usa Canada Pharmacy canadian family pharmacy canadianpharmacy.pro
http://pharmadoc.pro/# Pharmacie en ligne pas cher
Pharmacie en ligne fiable
http://levitrasansordonnance.pro/# Pharmacie en ligne fiable
Wohh precisely what I was searching for, regards for putting up.
My website: порно русский массаж
Pharmacie en ligne livraison 24h: kamagra en ligne – Pharmacie en ligne livraison 24h
Acheter mГ©dicaments sans ordonnance sur internet levitra generique prix en pharmacie pharmacie ouverte 24/24
Acheter mГ©dicaments sans ordonnance sur internet: levitra generique sites surs – acheter medicament a l etranger sans ordonnance
Pharmacie en ligne fiable Cialis sans ordonnance 24h Pharmacie en ligne fiable
http://cialissansordonnance.shop/# acheter médicaments à l’étranger
Thank you ever so for you blog. Really looking forward to read more.
My website: секс с изнасилованием
pharmacie ouverte: Pharmacie en ligne fiable – acheter medicament a l etranger sans ordonnance
Viagra gГ©nГ©rique sans ordonnance en pharmacie viagrasansordonnance.pro Viagra homme prix en pharmacie sans ordonnance
https://viagrasansordonnance.pro/# Sildénafil 100 mg prix en pharmacie en France
https://cialissansordonnance.shop/# Pharmacie en ligne fiable
acheter medicament a l etranger sans ordonnance
Thanks for sharing, this is a fantastic blog post.Really thank you! Much obliged.
My website: порно препод
pharmacie ouverte: kamagra gel – Pharmacie en ligne sans ordonnance
https://acheterkamagra.pro/# Pharmacie en ligne France
pharmacie ouverte 24/24 PharmaDoc pharmacie ouverte
Sildenafil teva 100 mg sans ordonnance: Meilleur Viagra sans ordonnance 24h – Viagra sans ordonnance 24h
https://acheterkamagra.pro/# Pharmacies en ligne certifiées
Pharmacie en ligne fiable: pharmacie en ligne pas cher – Pharmacie en ligne pas cher
http://viagrasansordonnance.pro/# Sildénafil Teva 100 mg acheter
https://cialissansordonnance.shop/# Pharmacie en ligne livraison gratuite
acheter medicament a l etranger sans ordonnance
Viagra gГ©nГ©rique sans ordonnance en pharmacie: viagrasansordonnance.pro – Viagra sans ordonnance 24h Amazon
Pharmacie en ligne France Levitra pharmacie en ligne Pharmacie en ligne sans ordonnance
Definitely, what a great blog and revealing posts, I definitely will bookmark your site. Best Regards!
My website: порно студентки русское
buy zithromax 1000mg online: zithromax online paypal – zithromax 500
https://ivermectin.store/# purchase stromectol
ivermectin 0.5 lotion india: stromectol medication – ivermectin 1%
Definitely, what a great blog and revealing posts, I definitely will bookmark your site. Best Regards!
My website: наше порно
stromectol for head lice stromectol brand where can i buy stromectol
https://prednisonetablets.shop/# buy prednisone tablets uk
amoxicillin 500mg buy online uk: amoxicillin 500mg for sale uk – order amoxicillin online no prescription
http://amoxicillin.bid/# amoxicillin 50 mg tablets
prednisone buy online nz buy prednisone 5mg canada buy prednisone 5mg canada
how to get clomid without insurance: where can i buy clomid without a prescription – can i order cheap clomid no prescription
https://azithromycin.bid/# zithromax 250 price
ivermectin tablets order buy ivermectin cream for humans stromectol 3mg tablets
amoxicillin from canada: amoxicillin 500mg over the counter – prescription for amoxicillin
http://prednisonetablets.shop/# prednisone 50 mg buy
https://azithromycin.bid/# zithromax 500mg price
54 prednisone order prednisone online no prescription prednisone cost in india
can i get generic clomid without prescription: where to buy generic clomid without rx – where to buy cheap clomid online
http://prednisonetablets.shop/# prednisone 20mg nz
where can i buy zithromax capsules: order zithromax over the counter – zithromax canadian pharmacy
buy amoxicillin online no prescription: amoxicillin pharmacy price – buy amoxicillin 500mg online
generic prednisone cost prednisone 10 mg canada 100 mg prednisone daily
https://azithromycin.bid/# zithromax tablets
purchase prednisone canada: prednisone 60 mg tablet – buy prednisone online from canada
zithromax for sale 500 mg zithromax order online uk buy zithromax 1000mg online
https://ivermectin.store/# ivermectin 9mg
can i buy clomid online: generic clomid without a prescription – how to get cheap clomid pills
stromectol cost: ivermectin 0.2mg – cost of ivermectin cream
http://prednisonetablets.shop/# prednisone 20mg prescription cost
http://azithromycin.bid/# azithromycin zithromax
amoxicillin price without insurance prescription for amoxicillin amoxicillin online canada
cheap amoxicillin 500mg: amoxicillin 500mg buy online canada – amoxicillin medicine
https://azithromycin.bid/# generic zithromax 500mg india
Your article helped me a lot, is there any more related content? Thanks! https://accounts.binance.com/ka-GE/register-person?ref=GJY4VW8W
buying prescription drugs in mexico online: Online Pharmacies in Mexico – pharmacies in mexico that ship to usa mexicanpharm.shop
buying prescription drugs in mexico: mexican pharmaceuticals online – mexican drugstore online mexicanpharm.shop
https://canadianpharm.store/# vipps canadian pharmacy canadianpharm.store
top online pharmacy india: international medicine delivery from india – top 10 online pharmacy in india indianpharm.store
buying prescription drugs in mexico online Online Mexican pharmacy mexico pharmacy mexicanpharm.shop
canadian pharmacy near me: Canada Pharmacy online – canadian drugstore online canadianpharm.store
http://mexicanpharm.shop/# purple pharmacy mexico price list mexicanpharm.shop
https://canadianpharm.store/# canadian mail order pharmacy canadianpharm.store
india online pharmacy international medicine delivery from india indian pharmacy paypal indianpharm.store
indianpharmacy com: Indian pharmacy to USA – cheapest online pharmacy india indianpharm.store
https://mexicanpharm.shop/# best mexican online pharmacies mexicanpharm.shop
mexican border pharmacies shipping to usa: Online Mexican pharmacy – buying prescription drugs in mexico mexicanpharm.shop
cheapest online pharmacy india: Indian pharmacy to USA – buy medicines online in india indianpharm.store
reputable mexican pharmacies online Online Mexican pharmacy mexican mail order pharmacies mexicanpharm.shop
canadian pharmacy reviews: canadian mail order pharmacy – canadian pharmacy online reviews canadianpharm.store
https://indianpharm.store/# india pharmacy mail order indianpharm.store
world pharmacy india order medicine from india to usa indianpharmacy com indianpharm.store
indian pharmacy online: order medicine from india to usa – online shopping pharmacy india indianpharm.store
https://canadianpharm.store/# canadian pharmacy india canadianpharm.store
india pharmacy mail order: international medicine delivery from india – Online medicine home delivery indianpharm.store
mexico drug stores pharmacies: Online Pharmacies in Mexico – mexico pharmacies prescription drugs mexicanpharm.shop
https://indianpharm.store/# online shopping pharmacy india indianpharm.store
safe canadian pharmacies Best Canadian online pharmacy canada pharmacy online legit canadianpharm.store
https://indianpharm.store/# top 10 pharmacies in india indianpharm.store
best online canadian pharmacy: Canada Pharmacy online – ed meds online canada canadianpharm.store
mexican online pharmacies prescription drugs: pharmacies in mexico that ship to usa – best online pharmacies in mexico mexicanpharm.shop
http://indianpharm.store/# india online pharmacy indianpharm.store
pet meds without vet prescription canada Canadian Pharmacy best mail order pharmacy canada canadianpharm.store
canadian discount pharmacy: Canada Pharmacy online – safe canadian pharmacy canadianpharm.store
http://canadianpharm.store/# my canadian pharmacy reviews canadianpharm.store
A round of applause for your article. Much thanks again.
My website: порно студентов
buying prescription drugs in mexico: Certified Pharmacy from Mexico – mexico drug stores pharmacies mexicanpharm.shop
canadian pharmacies compare: Pharmacies in Canada that ship to the US – canadian king pharmacy canadianpharm.store
mexico pharmacy Online Mexican pharmacy mexican drugstore online mexicanpharm.shop
https://canadianpharm.store/# global pharmacy canada canadianpharm.store
http://mexicanpharm.shop/# mexican online pharmacies prescription drugs mexicanpharm.shop
buying prescription drugs in mexico: Online Pharmacies in Mexico – buying from online mexican pharmacy mexicanpharm.shop
I’m extremely pleased to discover this website. I wanted to thank you for ones time just for this fantastic read!
My website: порно с русскими студентками
reliable canadian pharmacy: Canadian International Pharmacy – safe online pharmacies in canada canadianpharm.store
п»їbest mexican online pharmacies Online Pharmacies in Mexico mexico drug stores pharmacies mexicanpharm.shop
https://mexicanpharm.shop/# mexican pharmaceuticals online mexicanpharm.shop
mexican rx online: Online Mexican pharmacy – mexico pharmacy mexicanpharm.shop
«Миссия и предназначение»
«Идея всей жизни!»
Миссия – «Что я делаю?» Нет.
Ценности – «Что для меня важно?» тоже нет.
Видение – «Куда я иду (хочу придти)?» уже ближе.
Миссия – стержень: вектор: который определяет смысл!
Вбивай в поиск: “опсуимолог” и получи актуальные контакты.
Ты же знаешь кто такой опсуимолог?
mexican drugstore online Online Pharmacies in Mexico п»їbest mexican online pharmacies mexicanpharm.shop
http://indianpharm.store/# indian pharmacy indianpharm.store
buy medicines online in india: order medicine from india to usa – indian pharmacy paypal indianpharm.store
buy prescription drugs from india: international medicine delivery from india – reputable indian pharmacies indianpharm.store
reputable indian pharmacies: international medicine delivery from india – reputable indian pharmacies indianpharm.store
A lot of blog writers nowadays yet just a few have blog posts worth spending time on reviewing.
My website: порно русские мамки анал
https://indianpharm.store/# india online pharmacy indianpharm.store
https://mexicanpharm.shop/# best online pharmacies in mexico mexicanpharm.shop
Muchos Gracias for your article.Really thank you! Cool.
My website: смотреть порно глубокая глотка
purple pharmacy mexico price list Certified Pharmacy from Mexico medication from mexico pharmacy mexicanpharm.shop
cheapest online pharmacy india: international medicine delivery from india – indian pharmacy indianpharm.store
reliable mexican pharmacy: online drugstore reviews – no prior prescription required pharmacy
Major thanks for the article post. Much thanks again.
My website: букаки
https://canadadrugs.pro/# pharmacies withour prescriptions
canada online pharmacy: mexican border pharmacies – cheap canadian pharmacy
canadian drug company safe online pharmacy discount drug store online shopping
I am incessantly thought about this, thanks for posting.
My website: лижет очко подруге
top canadian pharmacies: prescription drugs canada – canada pharmacy world
http://canadadrugs.pro/# online drugstore reviews
best rated canadian pharmacy: foreign online pharmacy – discount canadian pharmacy
prescriptions canada canadian prescription drugs pharmacy canada online
canada pharmaceuticals online: canadian pharcharmy – reliable online pharmacy
https://canadadrugs.pro/# pain meds online without doctor prescription
online canadian pharmaceutical companies: canadian pharmacy antibiotics – no prescription rx medicine
trusted canadian online pharmacy mexican drug pharmacy highest discount on medicines online
My website: порно казачка
http://canadadrugs.pro/# legitimate online pharmacy usa
nabp approved canadian pharmacies: best online pharmacies without prescription – viagra online canadian pharmacy
A round of applause for your article. Much thanks again.
My website: порно вылизал жопу
no prescription drugs canada: recommended canadian pharmacies – online prescriptions without script
non prescription canadian pharmacy canadian drug store viagra best canadian drugstore
online pharmacies: prescription drug prices – legitimate canadian internet pharmacies
https://canadadrugs.pro/# mexican online pharmacy
fda approved canadian online pharmacies: medicine prices – canada pharmacies without script
canadian pharmacy shop: canadian pharmacies without prescriptions – verified canadian pharmacies
http://canadadrugs.pro/# canadian prescriptions
canadian pharmacy antiobotics without perscription: canadian online pharmacies not requiring a prescription – mexico pharmacy order online
A lot of blog writers nowadays yet just a few have blog posts worth spending time on reviewing.
My website: транс ебет мужика
http://canadadrugs.pro/# reliable online drugstore
cheap canadian pharmacy: verified canadian pharmacy – canadian drug
https://canadadrugs.pro/# online pharmacies without an rx
no perscription pharmacy: online pharmacy store – online pharmacy no prescription
mexican drug pharmacy: mail order pharmacy canada – best online canadian pharmacy
legitimate canadian pharmacies online: canadian xanax – canadian online pharmacies ratings
This site definitely has all of the information I needed about this subject
My website: волосатые пизды женщин
https://canadadrugs.pro/# canadian pharmacy without a prescription
good online mexican pharmacy: canadian pharmacy price checker – canadian pharmacy azithromycin
http://canadadrugs.pro/# mexican pharmacies that ship
online pharmacy usa: top 10 mail order pharmacies – canadian world pharmacy
http://canadadrugs.pro/# medications without prescription
Ponto IPTV a melhor programacao de canais IPTV do Brasil, filmes, series, futebol
My website: анал очень больно
ed meds online without doctor prescription: prescription drugs online without doctor – viagra without doctor prescription
https://certifiedpharmacymexico.pro/# mexican rx online
viagra without doctor prescription: ed pills without doctor prescription – ed meds online without doctor prescription
canadian pharmacy meds reviews online canadian pharmacy reviews the canadian drugstore
https://edwithoutdoctorprescription.pro/# real viagra without a doctor prescription usa
http://edwithoutdoctorprescription.pro/# prescription meds without the prescriptions
generic ed drugs: best ed treatment pills – over the counter erectile dysfunction pills
ed meds online without prescription or membership generic cialis without a doctor prescription meds online without doctor prescription
Thanks for the complete information. You helped me.
http://edpill.cheap/# cheap ed drugs
buy medicines online in india best india pharmacy india pharmacy mail order
ed meds online without doctor prescription: cheap cialis – buy cheap prescription drugs online
buying prescription drugs in mexico: buying prescription drugs in mexico – purple pharmacy mexico price list
http://edwithoutdoctorprescription.pro/# mexican pharmacy without prescription
prescription drugs online without doctor cheap cialis prescription drugs online without doctor
http://certifiedpharmacymexico.pro/# mexican mail order pharmacies
best over the counter ed pills best ed pills online treatment for ed
https://canadianinternationalpharmacy.pro/# is canadian pharmacy legit
Online medicine order: п»їlegitimate online pharmacies india – reputable indian pharmacies
best online pharmacies in mexico: mexico pharmacies prescription drugs – mexico pharmacies prescription drugs
the best ed pills buy erection pills erection pills
Thanks-a-mundo for the post.Really thank you! Awesome.
My website: порно скрытно
http://medicinefromindia.store/# world pharmacy india
A lot of blog writers nowadays yet just a few have blog posts worth spending time on reviewing.
My website: голые маленькие сиськи
http://edpill.cheap/# cheap erectile dysfunction pills online
mexico pharmacies prescription drugs mexico drug stores pharmacies medication from mexico pharmacy
http://medicinefromindia.store/# india pharmacy
best online pharmacy india buy medicines online in india top online pharmacy india
ed pills gnc: ed pills comparison – medication for ed
http://medicinefromindia.store/# pharmacy website india
Thanks-a-mundo for the post.Really thank you! Awesome.
My website: русский секс в попу
best india pharmacy buy medicines online in india mail order pharmacy india
https://medicinefromindia.store/# world pharmacy india
mexican mail order pharmacies mexican pharmacy buying prescription drugs in mexico online
https://canadianinternationalpharmacy.pro/# canadian pharmacy reviews
http://medicinefromindia.store/# online pharmacy india
indian pharmacies safe indian pharmacy paypal top 10 online pharmacy in india
pharmacies in mexico that ship to usa: pharmacies in mexico that ship to usa – buying prescription drugs in mexico
I got what you intend,bookmarked, very decent website.
My website: жена трахает страпоном
https://medicinefromindia.store/# india pharmacy mail order
mexican drugstore online п»їbest mexican online pharmacies п»їbest mexican online pharmacies
https://edpill.cheap/# cure ed
mexican pharmaceuticals online mexican pharmaceuticals online mexican border pharmacies shipping to usa
Muchos Gracias for your article.Really thank you! Cool.
My website: порно милф 50
https://edwithoutdoctorprescription.pro/# ed prescription drugs
levitra without a doctor prescription: cialis without a doctor prescription canada – prescription drugs online
best erectile dysfunction pills men’s ed pills cheap erectile dysfunction pills
http://edwithoutdoctorprescription.pro/# viagra without a doctor prescription
Wohh precisely what I was searching for, regards for putting up.
My website: русское порно измена мужу
best non prescription ed pills generic cialis without a doctor prescription ed meds online without doctor prescription
http://certifiedpharmacymexico.pro/# mexico drug stores pharmacies
http://medicinefromindia.store/# reputable indian pharmacies
onlineSlot machines Plus, find out how members of the Winners Club get even MORE benefits when playing their favorite games. Earn points for slot play, dining, and hotel, plus receive exclusive offers, additional perks, and so much more. Join the club! Akwesasne Mohawk Casino has delayed its reopening until August 28 due to eight new cases of COVID-19. The planned opening date was originally August 1. About The Akwesasne Mohawk Casino Resort Gambling in Cornwall, ON Just over an hour South-West from Montreal, Cornwall sits on the North bank of the St Lawrence river in a part of Ontario famous for food, farming and festivals, with the famous Ribfest showcasing some of the best BBQ food every summer. Cornwall is also rightContinue Reading Everyone’s Favorite Italian Classics are coming to MGM Springfield.
http://lunagaelle.blogspot.com/2007/04/micro-expressions.html
If you enjoy slots from WMS, IGT, Aristocrat and others, then you’ll now find many of them at online casinos. In a future regulated online casino market in Colorado, you can expect these to be listed among titles created especially for online use. Slots work well on casino apps, fitting perfectly onto your smartphone screen when you flip into landscape view. There are slots with progressive jackpots, interactive bonus games and games themed on big movie and TV franchises online. We suggest you check out Luckyland Slots’ sister casino or the sites like Fortune Coins, and High 5 Casino as they offer games like Chumba. BetOnline is a traditional online casino known for its Sportsbook, but it also provides thrilling casino slot games, live dealer games, and video poker. You can play slot machine games and many other games at this online casino.
https://canadianinternationalpharmacy.pro/# canadian neighbor pharmacy
mail order pharmacy india online shopping pharmacy india indianpharmacy com
I gotta favorite this site it seems very beneficial handy
My website: Порно подростков
drugs for ed: erectile dysfunction medications – mens ed pills
http://certifiedpharmacymexico.pro/# mexico pharmacy
mexican drugstore online mexican pharmacy mexico drug stores pharmacies
symbicort inhaler for asthma 160/4.5 – Medication safety audits should assess the adequacy of medication storage practices to prevent degradation.
http://certifiedpharmacymexico.pro/# mexican pharmacy
Respect to post author, some fantastic information
My website: секс сцены подростков
prescription meds without the prescriptions cheap cialis real viagra without a doctor prescription usa
https://mexicanph.com/# mexican online pharmacies prescription drugs
mexico drug stores pharmacies
buying from online mexican pharmacy purple pharmacy mexico price list mexican pharmaceuticals online
medication from mexico pharmacy best online pharmacies in mexico buying prescription drugs in mexico
medication from mexico pharmacy mexico pharmacy mexico drug stores pharmacies
buying from online mexican pharmacy mexican online pharmacies prescription drugs medicine in mexico pharmacies
п»їbest mexican online pharmacies best online pharmacies in mexico mexican border pharmacies shipping to usa
Respect to post author, some fantastic information
My website: секс жмж частное
reputable mexican pharmacies online mexican rx online pharmacies in mexico that ship to usa
mexican pharmacy buying prescription drugs in mexico online pharmacies in mexico that ship to usa
http://mexicanph.shop/# buying prescription drugs in mexico
medication from mexico pharmacy
mexico drug stores pharmacies mexican mail order pharmacies buying prescription drugs in mexico
mexican drugstore online mexico drug stores pharmacies buying prescription drugs in mexico online
mexican border pharmacies shipping to usa buying prescription drugs in mexico purple pharmacy mexico price list
https://mexicanph.shop/# mexican online pharmacies prescription drugs
buying prescription drugs in mexico
reputable mexican pharmacies online mexican pharmacy medication from mexico pharmacy
mexican border pharmacies shipping to usa mexico drug stores pharmacies medication from mexico pharmacy
mexico pharmacies prescription drugs purple pharmacy mexico price list purple pharmacy mexico price list
играть в казино на рубли
buying prescription drugs in mexico mexican drugstore online mexican mail order pharmacies
buying prescription drugs in mexico mexican mail order pharmacies mexican drugstore online
A round of applause for your article. Much thanks again.
My website: смотреть групповой анал
buying prescription drugs in mexico online reputable mexican pharmacies online pharmacies in mexico that ship to usa
buying prescription drugs in mexico mexico drug stores pharmacies pharmacies in mexico that ship to usa
mexico drug stores pharmacies п»їbest mexican online pharmacies mexican border pharmacies shipping to usa
mexican border pharmacies shipping to usa mexican mail order pharmacies reputable mexican pharmacies online
mexico pharmacy mexico pharmacy purple pharmacy mexico price list
This site definitely has all of the information I needed about this subject
My website: порно групповой минет
mexico pharmacy mexican pharmaceuticals online purple pharmacy mexico price list
https://mexicanph.com/# mexican border pharmacies shipping to usa
buying prescription drugs in mexico
A round of applause for your article. Much thanks again.
My website: глубокий минет
medicine in mexico pharmacies mexico drug stores pharmacies reputable mexican pharmacies online
medication from mexico pharmacy mexico pharmacies prescription drugs mexican drugstore online
buying from online mexican pharmacy mexican rx online п»їbest mexican online pharmacies
mexican rx online medicine in mexico pharmacies best online pharmacies in mexico
п»їbest mexican online pharmacies mexican drugstore online mexico drug stores pharmacies
mexican rx online mexican online pharmacies prescription drugs purple pharmacy mexico price list
medicine in mexico pharmacies mexican mail order pharmacies mexican pharmacy
mexican online pharmacies prescription drugs mexico drug stores pharmacies buying prescription drugs in mexico online
https://mexicanph.shop/# buying prescription drugs in mexico online
buying from online mexican pharmacy
Major thanks for the article post. Much thanks again.
My website: домашний анал молодых
mexican drugstore online mexican online pharmacies prescription drugs mexico pharmacies prescription drugs
reputable mexican pharmacies online mexican border pharmacies shipping to usa mexican mail order pharmacies
buying prescription drugs in mexico online mexican pharmacy best online pharmacies in mexico
mexico drug stores pharmacies mexican drugstore online mexico drug stores pharmacies
Thanks for sharing, this is a fantastic blog post.Really thank you! Much obliged.
My website: жена в черных чулках
гама казино
reputable mexican pharmacies online mexican pharmaceuticals online buying prescription drugs in mexico online
mexican mail order pharmacies mexican mail order pharmacies buying from online mexican pharmacy
pharmacies in mexico that ship to usa buying from online mexican pharmacy mexican border pharmacies shipping to usa
mexico drug stores pharmacies п»їbest mexican online pharmacies mexican rx online
medication from mexico pharmacy buying from online mexican pharmacy buying prescription drugs in mexico online
Muchos Gracias for your article.Really thank you! Cool.
My website: казахское порно сливы
purple pharmacy mexico price list pharmacies in mexico that ship to usa mexican drugstore online
mexican border pharmacies shipping to usa mexico pharmacy mexican online pharmacies prescription drugs
mexico drug stores pharmacies mexican drugstore online mexico pharmacy
I’m extremely pleased to discover this website. I wanted to thank you for ones time just for this fantastic read!
My website: пожилые лесбиянки
buying prescription drugs in mexico online mexican rx online mexican rx online
mexico pharmacies prescription drugs medication from mexico pharmacy mexican online pharmacies prescription drugs
You can’t get a cup of tea big sufficient or a book long enough to suit me.
best online pharmacies in mexico mexico pharmacy buying prescription drugs in mexico online
http://mexicanph.com/# buying prescription drugs in mexico online
mexico pharmacies prescription drugs
buying from online mexican pharmacy best online pharmacies in mexico mexico drug stores pharmacies
buying prescription drugs in mexico best mexican online pharmacies mexican rx online
mexico pharmacies prescription drugs mexican online pharmacies prescription drugs mexico pharmacies prescription drugs
medicine in mexico pharmacies mexican border pharmacies shipping to usa medication from mexico pharmacy
I am incessantly thought about this, thanks for posting.
My website: порно секс студентки
mexico pharmacies prescription drugs best mexican online pharmacies mexican rx online
medicine in mexico pharmacies mexican border pharmacies shipping to usa mexico pharmacy
buying from online mexican pharmacy mexican border pharmacies shipping to usa mexican border pharmacies shipping to usa
mexican rx online medication from mexico pharmacy best online pharmacies in mexico
purple pharmacy mexico price list mexico pharmacies prescription drugs mexican rx online
medicine in mexico pharmacies best online pharmacies in mexico mexican mail order pharmacies
Definitely, what a great blog and revealing posts, I definitely will bookmark your site. Best Regards!
My website: жесткое порно бесплатно
https://mexicanph.shop/# mexico drug stores pharmacies
mexican border pharmacies shipping to usa
medication from mexico pharmacy pharmacies in mexico that ship to usa mexican online pharmacies prescription drugs
mexico drug stores pharmacies mexico drug stores pharmacies mexican mail order pharmacies
purple pharmacy mexico price list mexican pharmaceuticals online mexico drug stores pharmacies
mexican drugstore online mexican rx online mexico drug stores pharmacies
mexican online pharmacies prescription drugs best mexican online pharmacies buying prescription drugs in mexico online
I reckon something truly special in this website.
My website: трахают в жопу смотреть
mexican mail order pharmacies mexican rx online pharmacies in mexico that ship to usa
buying prescription drugs in mexico mexican online pharmacies prescription drugs mexico drug stores pharmacies
stromectol buy uk: ivermectin 1 cream 45gm – ivermectin 4 tablets price
http://furosemide.guru/# lasix 40mg
http://buyprednisone.store/# prednisone online australia
generic amoxicillin cost amoxil pharmacy where to buy amoxicillin 500mg without prescription
prednisone 20 mg purchase: how can i get prednisone – prednisone 20mg online without prescription
https://furosemide.guru/# lasix 20 mg
http://furosemide.guru/# lasix pills
stromectol for sale: stromectol price us – stromectol price us
http://furosemide.guru/# lasix pills
ivermectin 3 mg ivermectin cost australia stromectol covid
I reckon something truly special in this website.
My website: ебля от первого лица
ivermectin price comparison: ivermectin nz – ivermectin 50 mg
http://buyprednisone.store/# non prescription prednisone 20mg
generic prinivil lisinopril in usa lisinopril 20 mg buy
ivermectin oral 0 8: ivermectin price uk – ivermectin buy
http://buyprednisone.store/# prednisone 2.5 mg cost
Thanks for sharing, this is a fantastic blog post.Really thank you! Much obliged.
My website: секс по дружбе
https://buyprednisone.store/# prednisone brand name india
ivermectin 4 tablets price stromectol tablets uk stromectol generic
prednisone medication: prednisone 20mg – prednisone generic brand name
Respect to post author, some fantastic information
My website: arabian porno
http://lisinopril.top/# prescription drug lisinopril
lisinopril 20mg: lisinopril tablet – order lisinopril online
https://amoxil.cheap/# amoxicillin order online
prednisone 20mg 5mg prednisone prednisone 10 mg
lasix online: furosemida – buy lasix online
https://amoxil.cheap/# order amoxicillin no prescription
https://amoxil.cheap/# buy amoxicillin without prescription
http://buyprednisone.store/# prednisone buy no prescription
where can i purchase lisinopril lisinopril online cheap lisinopril
buy amoxicillin without prescription: azithromycin amoxicillin – amoxicillin 30 capsules price
http://stromectol.fun/# stromectol uk
lisinopril 80mg tablet: generic lisinopril 10 mg – lisinopril 20g
http://lisinopril.top/# lisinopril without an rx
cost of amoxicillin 875 mg amoxicillin cephalexin buy amoxicillin 500mg canada
order prednisone online no prescription: where can i order prednisone 20mg – prednisone cost 10mg
http://buyprednisone.store/# prednisone prescription for sale
steroids prednisone for sale: prednisone 50 mg coupon – prednisone 20 mg purchase
https://lisinopril.top/# zestoretic 20
over the counter amoxicillin buy amoxicillin 500mg capsules uk amoxil pharmacy
https://lisinopril.top/# lisinopril 2.5 mg
https://buyprednisone.store/# prednisone tablets india
ivermectin buy online: buy ivermectin cream for humans – stromectol 0.5 mg
https://buyprednisone.store/# how can i get prednisone
where to buy prednisone uk 80 mg prednisone daily order prednisone with mastercard debit
5 prednisone in mexico: prednisone buy canada – prednisone pill 20 mg
http://furosemide.guru/# lasix dosage
buy amoxicillin online cheap: amoxicillin 500 – order amoxicillin no prescription
https://stromectol.fun/# ivermectin cost in usa
amoxicillin discount amoxicillin 500mg amoxicillin 500mg price
https://stromectol.fun/# ivermectin 90 mg
https://lisinopril.top/# lisinopril 419
prednisone 20mg price in india: prednisone 20mg online without prescription – prednisone no rx
I do not know if it’s just me or if everyone else encountering issues with your site. It appears as if some of the written text within your content are running off the screen. Can somebody else please comment and let me know if this is happening to them as well? This might be a problem with my browser because I’ve had this happen before. Cheers
https://buyprednisone.store/# 40 mg daily prednisone
furosemide dogs
I have seen wonderful websites and I have caught not so great websites. This site is very informative in many ways and certainloy ranks in the former category. Really appreciate the information your providing use avid readers!
can i purchase prednisone without a prescription prednisone 5mg over the counter prednisone medication
how much is prednisone 10 mg: buy prednisone online without a prescription – buy prednisone 40 mg
Sup, this is fantastic guide. I absolutely enjoyed reading. However there are a lot of off topic comments. I seriously recommend you to delete or something like that. That’s only my estimation. All the best!
http://stromectol.fun/# stromectol ivermectin tablets
i have a brother that is autistic and we love him so much and gave all of our support on him;
I appreciate, cause I found just what I was looking for. You’ve ended my four day long hunt! God Bless you man. Have a nice day. Bye
buy prednisone online from canada: prednisone 1mg purchase – prednisone price canada
https://furosemide.guru/# generic lasix
hey all, I used to be simply checking out this weblog and I really admire the idea of the article, and have nothing to do, so if anybody want to to have an engrossing convo about it, please contact me on AIM, my identify is heather smith
buy lisinopril 20 mg online canada canadian lisinopril 10 mg lisinopril 40 mg india
You have a very nice layout for your blog, i want it to use on my site too ,
http://amoxil.cheap/# amoxicillin 500mg price in canada
Can I merely say thats a relief to find someone that really knows what theyre talking about over the internet. You certainly know how to bring an issue to light to make it critical. The diet need to ought to see this and understand this side from the story. I cant believe youre less well-liked because you undoubtedly provide the gift.
can i purchase amoxicillin online: amoxicillin 250 mg – amoxicillin 775 mg
https://lisinopril.top/# lisinopril 1.25 mg
It’s the best time to make some plans for the future and it is time to be happy. I’ve read this post and if I could I wish to suggest you some interesting things or advice. Maybe you can write next articles referring to this article. I wish to read even more things about it!
https://lisinopril.top/# 40 mg lisinopril
prednisone without prescription 10mg prednisone without prescription prednisone medicine
cost of amoxicillin prescription: amoxicillin 500 mg for sale – amoxicillin 250 mg capsule
http://furosemide.guru/# generic lasix
https://lisinopril.top/# zestoretic price
lasix 100 mg tablet: Buy Furosemide – lasix generic name
Youre so cool! I dont suppose Ive read anything such as this before. So nice to get somebody with some original thoughts on this subject. realy we appreciate you starting this up. this fabulous website are some things that is required on the internet, somebody with a little originality. beneficial work for bringing a new challenge on the world wide web!
ivermectin 0.5 lotion india ivermectin 0.5% ivermectin lotion
https://furosemide.guru/# lasix online
gabapentin dosage for nerve pain
My wife and i ended up being absolutely fulfilled Chris managed to carry out his studies from your ideas he had out of the web pages. It is now and again perplexing to just happen to be releasing facts that many many others may have been selling. Therefore we remember we now have the blog owner to thank because of that. All of the explanations you made, the easy blog menu, the relationships you can help to engender – it’s got mostly spectacular, and it’s really assisting our son and us reason why the theme is exciting, which is very indispensable. Many thanks for the whole thing!
lisinopril 40 mg tablet: prinivil 25mg – lisinopril 10 mg online no prescription
I have learned newer and more effective things by means of your blog. One other thing I’d like to say is that newer personal computer operating systems are inclined to allow extra memory to get used, but they likewise demand more memory simply to function. If a person’s computer can’t handle extra memory and also the newest computer software requires that memory increase, it could be the time to buy a new Laptop. Thanks
http://furosemide.guru/# lasix generic name
Youre so cool! I dont suppose Ive read anything such as this before. So nice to get somebody by original ideas on this subject. realy thank you for starting this up. this amazing site is a thing that is needed on-line, a person with a bit of originality. valuable task for bringing new stuff on the world wide web!
https://furosemide.guru/# furosemide 100 mg
https://stromectol.fun/# stromectol where to buy
amoxicillin 500mg buy online canada: buy amoxicillin without prescription – buying amoxicillin in mexico
ivermectin brand ivermectin 5 buy ivermectin uk
https://lisinopril.top/# 2 lisinopril
prednisone buy canada: buy prednisone online without a script – buy prednisone online without a prescription
gabapentin alcohol
http://indianph.xyz/# indian pharmacies safe
buy prescription drugs from india
indian pharmacies safe world pharmacy india cheapest online pharmacy india
http://indianph.xyz/# best online pharmacy india
top 10 online pharmacy in india
http://indianph.com/# reputable indian pharmacies
india pharmacy mail order
https://indianph.com/# indian pharmacies safe
https://indianph.com/# indianpharmacy com
india online pharmacy
http://indianph.xyz/# reputable indian pharmacies
reputable indian pharmacies
top 10 pharmacies in india top online pharmacy india online shopping pharmacy india
https://indianph.com/# online shopping pharmacy india
indian pharmacies safe
http://indianph.xyz/# india pharmacy mail order
india pharmacy mail order
Online medicine order online pharmacy india world pharmacy india
http://indianph.com/# indian pharmacies safe
diflucan prescription australia: diflucan india – diflucan 150mg
https://cipro.guru/# cipro online no prescription in the usa
п»їcipro generic ciprofloxacin generic where can i buy cipro online
http://cytotec24.shop/# buy cytotec online fast delivery
Abortion pills online: buy cytotec in usa – Cytotec 200mcg price
http://diflucan.pro/# diflucan over the counter nz
is nolvadex legal nolvadex generic tamoxifen hot flashes
http://doxycycline.auction/# price of doxycycline
http://doxycycline.auction/# doxycycline hydrochloride 100mg
doxycycline hyclate 100 mg cap: how to order doxycycline – 100mg doxycycline
http://diflucan.pro/# diflucan gel
where can i buy cipro online buy ciprofloxacin over the counter buy cipro
http://doxycycline.auction/# doxycycline generic
diflucan canadian pharmacy: diflucan fluconazole – diflucan brand name 300mg
п»їcytotec pills online: order cytotec online – buy cytotec pills online cheap
http://cytotec24.com/# cytotec online
ciprofloxacin 500mg buy online cipro online no prescription in the usa buy cipro cheap
http://cipro.guru/# ciprofloxacin mail online
https://diflucan.pro/# diflucan online australia
https://nolvadex.guru/# nolvadex estrogen blocker
cytotec pills buy online cytotec abortion pill Abortion pills online
http://doxycycline.auction/# doxycycline generic
http://cipro.guru/# cipro online no prescription in the usa
https://cipro.guru/# buy ciprofloxacin
buy doxycycline without prescription price of doxycycline doxycycline 150 mg
http://cipro.guru/# buy cipro online canada
http://doxycycline.auction/# doxycycline monohydrate
diflucan rx online diflucan 200 mg over the counter diflucan buy in usa
http://cytotec24.com/# buy cytotec
https://doxycycline.auction/# buy doxycycline
Latina girl has dirty games for you lots of fun in pvt
http://evaelfie.pro/# eva elfie video
Sweetie Fox modeli: Sweetie Fox – swetie fox
http://evaelfie.pro/# eva elfie video
http://angelawhite.pro/# Angela White filmleri
eva elfie: eva elfie izle – eva elfie filmleri
http://angelawhite.pro/# Angela Beyaz modeli
http://evaelfie.pro/# eva elfie filmleri
http://evaelfie.pro/# eva elfie
lana rhoades modeli: lana rhodes – lana rhoades video
http://evaelfie.pro/# eva elfie filmleri
http://angelawhite.pro/# Angela Beyaz modeli
https://sweetiefox.online/# Sweetie Fox izle
lana rhoades: lana rhoades izle – lana rhoades video
http://abelladanger.online/# abella danger izle
https://abelladanger.online/# abella danger video
http://angelawhite.pro/# Angela White video
Sweetie Fox modeli: Sweetie Fox modeli – sweety fox
https://lanarhoades.fun/# lana rhoades izle
http://evaelfie.pro/# eva elfie
eva elfie modeli: eva elfie filmleri – eva elfie izle
https://sweetiefox.online/# Sweetie Fox modeli
http://abelladanger.online/# abella danger izle
http://lanarhoades.fun/# lana rhodes
https://evaelfie.pro/# eva elfie izle
eva elfie filmleri: eva elfie izle – eva elfie
http://lanarhoades.fun/# lana rhoades
lana rhoades: lana rhoades modeli – lana rhoades izle
http://lanarhoades.fun/# lana rhoades izle
https://abelladanger.online/# Abella Danger
http://angelawhite.pro/# ?????? ????
eva elfie: eva elfie – eva elfie filmleri
http://abelladanger.online/# Abella Danger
https://evaelfie.pro/# eva elfie izle
http://evaelfie.pro/# eva elfie video
Angela White: abella danger izle – abella danger izle
http://abelladanger.online/# Abella Danger
https://lanarhoades.fun/# lana rhodes
https://sweetiefox.online/# sweety fox
http://angelawhite.pro/# Angela White filmleri
Sweetie Fox filmleri: Sweetie Fox – Sweetie Fox video
http://sweetiefox.online/# Sweetie Fox izle
http://evaelfie.pro/# eva elfie modeli
eva elfie: eva elfie – eva elfie video
http://abelladanger.online/# abella danger izle
eva elfie izle: eva elfie – eva elfie filmleri
https://evaelfie.pro/# eva elfie modeli
eva elfie new video: eva elfie full videos – eva elfie full video
https://evaelfie.site/# eva elfie
https://evaelfie.site/# eva elfie hd
mia malkova videos: mia malkova videos – mia malkova photos
http://evaelfie.site/# eva elfie hot
completely free dating sites: http://evaelfie.site/# eva elfie new video
lana rhoades solo: lana rhoades hot – lana rhoades videos
sweetie fox full: ph sweetie fox – sweetie fox new
https://lanarhoades.pro/# lana rhoades boyfriend
mia malkova full video: mia malkova photos – mia malkova hd
plenty of fish dating site: https://lanarhoades.pro/# lana rhoades pics
https://evaelfie.site/# eva elfie
mia malkova full video: mia malkova new video – mia malkova
http://lanarhoades.pro/# lana rhoades pics
sweetie fox full: fox sweetie – sweetie fox full video
free chat sites dating: http://evaelfie.site/# eva elfie new video
ph sweetie fox: sweetie fox cosplay – sweetie fox
sweetie fox: sweetie fox cosplay – sweetie fox full
http://sweetiefox.pro/# sweetie fox full video
lana rhoades solo: lana rhoades boyfriend – lana rhoades solo
http://evaelfie.site/# eva elfie full video
dating gmail germany: https://miamalkova.life/# mia malkova girl
eva elfie photo: eva elfie hot – eva elfie new video
mia malkova hd: mia malkova latest – mia malkova only fans
https://evaelfie.site/# eva elfie new videos
lana rhoades videos: lana rhoades – lana rhoades boyfriend
best dating websites online: http://evaelfie.site/# eva elfie full video
http://lanarhoades.pro/# lana rhoades
lana rhoades hot: lana rhoades full video – lana rhoades solo
mia malkova photos: mia malkova movie – mia malkova photos
sweetie fox new: ph sweetie fox – sweetie fox new
https://miamalkova.life/# mia malkova latest
meet me dating site free: http://miamalkova.life/# mia malkova
mia malkova: mia malkova full video – mia malkova videos
http://evaelfie.site/# eva elfie full video
eva elfie hd: eva elfie videos – eva elfie new videos
aviator ghana: aviator bet – aviator login
http://aviatorghana.pro/# aviator game
jogo de aposta: melhor jogo de aposta – aviator jogo de aposta
aviator ghana: aviator sportybet ghana – aviator game
https://aviatoroyunu.pro/# aviator oyna
aviator sinyal hilesi: aviator oyunu – pin up aviator
http://aviatormocambique.site/# como jogar aviator
aviator game online: play aviator – aviator game
pin-up cassino: pin-up casino login – pin-up casino
http://aviatoroyunu.pro/# pin up aviator
aviator game: aviator jogo – aviator game
https://pinupcassino.pro/# pin-up casino login
aviator game online: aviator game – play aviator
aviator game: aviator – aviator game bet
aviator sportybet ghana: play aviator – aviator
depósito mínimo 1 real: aviator jogo de aposta – melhor jogo de aposta
melhor jogo de aposta: depósito mínimo 1 real – jogo de aposta
pin up aviator: aviator jogar – aviator game
aviator: aviator game bet – aviator game online
jogar aviator: aviator mz – jogar aviator
melhor jogo de aposta: ganhar dinheiro jogando – jogo de aposta online
aviator: aviator bet – aviator
zithromax 500 mg lowest price online: zithromax and pregnancy can you buy zithromax over the counter
depósito mínimo 1 real: site de apostas – jogo de aposta online
where to buy zithromax in canada – https://azithromycin.pro/zithromax-pregnancy.html where to buy zithromax in canada
aviator game: aviator bet – aviator betting game
aviator sportybet ghana: aviator bet – aviator bet
zithromax for sale 500 mg: will zithromax treat sinus infection where can i buy zithromax in canada
melhor jogo de aposta: aplicativo de aposta – aviator jogo de aposta
aviator: aviator – play aviator
vipps canadian pharmacy Canada pharmacy online pharmacy canada canadianpharm.store
mexico pharmacy: Medicines Mexico – mexico pharmacy mexicanpharm.shop
pharmacy website india: Online India pharmacy – india pharmacy indianpharm.store
canada online pharmacy: canadian pharmacy – canadian pharmacy mall canadianpharm.store
medicine in mexico pharmacies order online from a Mexican pharmacy mexican border pharmacies shipping to usa mexicanpharm.shop
http://mexicanpharm24.com/# mexican mail order pharmacies mexicanpharm.shop
https://mexicanpharm24.com/# buying prescription drugs in mexico mexicanpharm.shop
my canadian pharmacy review: International Pharmacy delivery – maple leaf pharmacy in canada canadianpharm.store
http://canadianpharmlk.com/# reliable canadian pharmacy canadianpharm.store
https://indianpharm24.com/# top online pharmacy india indianpharm.store
mexican rx online: mexican pharmacy – reputable mexican pharmacies online mexicanpharm.shop
https://mexicanpharm24.com/# mexico pharmacies prescription drugs mexicanpharm.shop
mexican border pharmacies shipping to usa: Medicines Mexico – buying prescription drugs in mexico online mexicanpharm.shop
indian pharmacy Online India pharmacy п»їlegitimate online pharmacies india indianpharm.store
https://mexicanpharm24.com/# best online pharmacies in mexico mexicanpharm.shop
https://canadianpharmlk.com/# reputable canadian pharmacy canadianpharm.store
online canadian pharmacy reviews: Canada pharmacy online – pharmacy wholesalers canada canadianpharm.store
http://mexicanpharm24.shop/# buying prescription drugs in mexico mexicanpharm.shop
https://mexicanpharm24.shop/# reputable mexican pharmacies online mexicanpharm.shop
india pharmacy mail order: online pharmacy usa – india online pharmacy indianpharm.store
http://mexicanpharm24.com/# buying prescription drugs in mexico mexicanpharm.shop
pharmacy rx world canada: My Canadian pharmacy – reputable canadian pharmacy canadianpharm.store
http://canadianpharmlk.com/# trustworthy canadian pharmacy canadianpharm.store
indian pharmacy online india pharmacy indianpharmacy com indianpharm.store
https://mexicanpharm24.com/# mexican border pharmacies shipping to usa mexicanpharm.shop
http://mexicanpharm24.shop/# mexico pharmacy mexicanpharm.shop
indianpharmacy com: buy prescription drugs from india – top 10 pharmacies in india indianpharm.store
http://canadianpharmlk.com/# canadianpharmacymeds canadianpharm.store
https://mexicanpharm24.com/# medicine in mexico pharmacies mexicanpharm.shop
https://mexicanpharm24.shop/# mexican drugstore online mexicanpharm.shop
india pharmacy mail order: Online medicine home delivery – india online pharmacy indianpharm.store
buy prednisone mexico: prednisone 10mg for sale – can i order prednisone
https://amoxilst.pro/# amoxicillin 500mg price canada
buying prednisone on line: prednisone 30 mg tablet – prednisone 60 mg daily
how to buy cheap clomid online: where can i buy clomid – can i order clomid without prescription
http://prednisonest.pro/# can you buy prednisone over the counter in canada
where can i buy generic clomid without insurance: buy generic clomid without dr prescription – can you buy generic clomid for sale
prednisone 10mg prices: what is prednisone – where can i get prednisone
cheap clomid without prescription how to get generic clomid no prescription can i purchase generic clomid without dr prescription
amoxil pharmacy: how to buy amoxycillin – buy cheap amoxicillin
buying generic clomid without a prescription: clomid success rate – where can i get generic clomid pills
http://clomidst.pro/# buying generic clomid tablets
amoxicillin generic: where can i buy amoxicillin online – buy amoxicillin 500mg online
amoxicillin discount coupon: amoxicillin for tooth infection – amoxicillin where to get
hydroxychloroquine 200 mg for rheumatoid arthritis, a medication originally developed to treat malaria, has found widespread use in managing autoimmune conditions due to its immunomodulatory properties. By inhibiting certain immune responses, Plaquenil helps alleviate symptoms such as joint pain and inflammation in diseases like lupus and rheumatoid arthritis. While its precise mechanism of action isn’t fully understood, its ability to dampen immune activity has made it a valuable tool in the treatment arsenal for autoimmune disorders.
http://clomidst.pro/# can you buy generic clomid price
how to get amoxicillin over the counter: amoxicillin 500mg price – amoxacillian without a percription
where buy clomid without a prescription: side effects of clomid – how can i get generic clomid price
prednisone 50 mg tablet canada prednisone tablets prednisone 50 mg tablet canada
http://amoxilst.pro/# buy amoxicillin 500mg online
how to get clomid without insurance: best days to take clomid for twins – where buy clomid online
generic clomid without prescription: online doctor to prescribe clomid – how to get generic clomid without insurance
http://amoxilst.pro/# amoxicillin discount
prednisone 20 mg: prednisone cost canada – prednisone without prescription 10mg
order prednisone 10 mg tablet: how to purchase prednisone online – prednisone for cheap
cheap ed medication: erectile dysfunction pills for sale – ed med online
http://pharmnoprescription.pro/# can you buy prescription drugs in canada
ed meds cheap: best online ed treatment – cheapest online ed meds
canadian pharmacy world coupon: online mexican pharmacy – legit non prescription pharmacies
buy erectile dysfunction medication: cheapest ed medication – best ed medication online
pharmacy no prescription required discount pharmacy canadian pharmacy without prescription
http://edpills.guru/# buy ed medication
cheap ed pills: pills for erectile dysfunction online – buy ed pills online
no prescription pharmacy paypal: pharmacy online – no prescription needed pharmacy
cheap ed pills: low cost ed pills – online ed medications
http://pharmnoprescription.pro/# online meds without prescription
pharmacy no prescription: canadian pharmacy non prescription – buy medications without a prescription
cheap ed medication: erectile dysfunction medication online – ed doctor online
pharmacy coupons: online pharmacy – cheap pharmacy no prescription
https://onlinepharmacy.cheap/# best no prescription pharmacy
ed pills for sale: ed online meds – pills for ed online
pharmacy discount coupons online pharmacy delivery online pharmacy discount code
international pharmacy no prescription: Online pharmacy USA – legal online pharmacy coupon code
order prescription from canada: mexico prescription drugs online – online medicine without prescription
canadian pharmacy discount coupon: online mexican pharmacy – offshore pharmacy no prescription
http://onlinepharmacy.cheap/# pharmacy coupons
canadian online pharmacy no prescription: Best online pharmacy – canadian prescription pharmacy
world pharmacy india: top online pharmacy india – online shopping pharmacy india
http://canadianpharm.guru/# rate canadian pharmacies
https://mexicanpharm.online/# mexican border pharmacies shipping to usa
buy drugs online without a prescription can you buy prescription drugs in canada п»їonline pharmacy no prescription needed
mexico pharmacy: best online pharmacies in mexico – best online pharmacies in mexico
https://indianpharm.shop/# reputable indian pharmacies
best canadian online pharmacy: reliable canadian pharmacy reviews – my canadian pharmacy
aripiprazole 10
canada pharmacy online no prescription: online pharmacy no prescriptions – canadian pharmacy online no prescription
http://pharmacynoprescription.pro/# best online prescription
legitimate canadian pharmacy: canadapharmacyonline – maple leaf pharmacy in canada
http://mexicanpharm.online/# buying prescription drugs in mexico online
pharmacies in mexico that ship to usa: mexican mail order pharmacies – mexico drug stores pharmacies
mexican drugstore online mexico drug stores pharmacies mexico pharmacies prescription drugs
online drugs without prescription: online drugs no prescription – can i buy prescription drugs in canada
https://mexicanpharm.online/# mexican pharmaceuticals online
online shopping pharmacy india: best online pharmacy india – online shopping pharmacy india
prescription canada: buy meds online without prescription – buy prescription drugs online without doctor
http://mexicanpharm.online/# mexican pharmacy
http://pharmacynoprescription.pro/# how to get a prescription in canada
cheapest online pharmacy india: top 10 online pharmacy in india – mail order pharmacy india
mexico drug stores pharmacies mexican drugstore online medicine in mexico pharmacies
https://canadianpharm.guru/# canadian pharmacy online ship to usa
prescription online canada: online meds without prescription – online doctor prescription canada
indian pharmacy paypal: top online pharmacy india – indian pharmacy
meds online no prescription: online pharmacy not requiring prescription – no prescription on line pharmacies
http://canadianpharm.guru/# canadian pharmacy checker
order prescription from canada: no prescription – buy drugs online without a prescription
mexico pharmacy: best online pharmacies in mexico – pharmacies in mexico that ship to usa
http://pharmacynoprescription.pro/# buying prescription medications online
online pharmacy india: mail order pharmacy india – Online medicine home delivery
http://pharmacynoprescription.pro/# indian pharmacy no prescription
mexican online pharmacies prescription drugs п»їbest mexican online pharmacies mexican drugstore online
prescription meds from canada: best website to buy prescription drugs – order medication without prescription
online pharmacies without prescriptions: mexico online pharmacy prescription drugs – no prescription drugs online
https://indianpharm.shop/# reputable indian online pharmacy
best mexican online pharmacies: mexican pharmaceuticals online – mexico pharmacies prescription drugs
india pharmacy: pharmacy website india – pharmacy website india
http://canadianpharm.guru/# online canadian pharmacy reviews
certified canadian pharmacy: canada pharmacy reviews – canadian pharmacy
https://indianpharm.shop/# best india pharmacy
prescription drugs canada: online pharmacy with prescription – indian pharmacy no prescription
http://mexicanpharm.online/# pharmacies in mexico that ship to usa
reputable mexican pharmacies online best online pharmacies in mexico mexican border pharmacies shipping to usa
non prescription online pharmacy india: canadian prescription drugstore reviews – buying online prescription drugs
reputable indian pharmacies: india pharmacy – india pharmacy
mexico pharmacies prescription drugs: mexico pharmacy – buying prescription drugs in mexico
Pharmacovigilance plays a crucial role in monitoring the safety of stromectol and detecting adverse drug reactions in real-world clinical settings. Healthcare providers are encouraged to report suspected adverse events associated with stromectol for sale to regulatory authorities and pharmacovigilance systems to facilitate the timely identification and management of safety concerns. This proactive approach helps ensure that the benefits of stromectol outweigh its risks and contributes to the continuous improvement of drug safety surveillance and regulatory oversight.
https://indianpharm.shop/# world pharmacy india
Pharmacovigilance plays a crucial role in monitoring the safety of stromectol and detecting adverse drug reactions in real-world clinical settings. Healthcare providers are encouraged to report suspected adverse events associated with buy stromectol to regulatory authorities and pharmacovigilance systems to facilitate the timely identification and management of safety concerns. This proactive approach helps ensure that the benefits of stromectol outweigh its risks and contributes to the continuous improvement of drug safety surveillance and regulatory oversight.
safe canadian pharmacies: canada discount pharmacy – canadian pharmacy online reviews
buying online prescription drugs: medication online without prescription – online pharmacy no prescription
http://pharmacynoprescription.pro/# no prescription online pharmacies
mexico drug stores pharmacies: mexican pharmacy – mexican rx online
http://pharmacynoprescription.pro/# no prescription canadian pharmacy
canadian pharmacy service reputable canadian online pharmacies canadian pharmacy online
pharmacy website india: buy medicines online in india – cheapest online pharmacy india
https://indianpharm.shop/# top 10 pharmacies in india
buying from online mexican pharmacy: mexican rx online – mexico drug stores pharmacies
https://slotsiteleri.guru/# deneme bonusu veren siteler
sweet bonanza free spin demo: sweet bonanza kazanma saatleri – sweet bonanza
http://pinupgiris.fun/# aviator pin up
https://aviatoroyna.bid/# aviator oyna 100 tl
aviator sinyal hilesi ucretsiz: aviator oyunu – aviator nas?l oynan?r
http://sweetbonanza.bid/# sweet bonanza demo oyna
gates of olympus slot: gates of olympus oyna demo – gates of olympus demo oyna
http://aviatoroyna.bid/# aviator sinyal hilesi ücretsiz
sweet bonanza demo oyna: sweet bonanza 90 tl – sweet bonanza mostbet
https://aviatoroyna.bid/# aviator oyunu
https://gatesofolympus.auction/# gates of olympus
aviator pin up: pin up casino indir – pin up casino giris
http://slotsiteleri.guru/# casino slot siteleri
aviator oyunu 20 tl: aviator oyna slot – ucak oyunu bahis aviator
aviator giris: aviator giris – aviator oyunu 20 tl
http://gatesofolympus.auction/# gate of olympus hile
https://aviatoroyna.bid/# aviator oyunu giris
gates of olympus oyna ucretsiz: gates of olympus demo free spin – gates of olympus 1000 demo
https://sweetbonanza.bid/# sweet bonanza free spin demo
stromectol over the counter, with its active ingredient ivermectin, has shown efficacy in controlling parasitic infections in captive exotic arthropods such as tarantulas and scorpions. By targeting parasites such as mites and worms, ivermectin contributes to the overall health and well-being of these arachnids kept in captivity for research or as pets.
https://sweetbonanza.bid/# sweet bonanza güncel
pin-up online: pin up giris – pin-up bonanza
aviator pin up: pin-up online – pin up bet
https://pinupgiris.fun/# pin-up bonanza
sweet bonanza 100 tl: sweet bonanza slot demo – sweet bonanza indir
https://sweetbonanza.bid/# sweet bonanza nasil oynanir
http://gatesofolympus.auction/# gates of olympus demo
pin up aviator: pin up guncel giris – pin up indir
http://aviatoroyna.bid/# aviator hilesi
2024 en iyi slot siteleri: en yeni slot siteleri – slot oyun siteleri
sweet bonanza free spin demo: sweet bonanza demo turkce – sweet bonanza indir
https://slotsiteleri.guru/# slot siteleri güvenilir
https://pinupgiris.fun/# pin up bet
mexico pharmacy mexico pharmacy mexico pharmacies prescription drugs
https://indianpharmacy.icu/# buy medicines online in india
purple pharmacy mexico price list Mexican Pharmacy Online mexican mail order pharmacies
mexican online pharmacies prescription drugs: mexican pharmacy – п»їbest mexican online pharmacies
canadian pharmacy review Large Selection of Medications buy prescription drugs from canada cheap
canadian world pharmacy: pills now even cheaper – canadian drug pharmacy
top 10 online pharmacy in india: Generic Medicine India to USA – top online pharmacy india
india online pharmacy Healthcare and medicines from India india pharmacy mail order
mexican drugstore online: mexico pharmacy – best online pharmacies in mexico
http://mexicanpharmacy.shop/# medication from mexico pharmacy
best canadian online pharmacy: Certified Canadian Pharmacy – canadian drug pharmacy
india pharmacy: Generic Medicine India to USA – indian pharmacy online
canadian pharmacy 24h com safe Prescription Drugs from Canada canadian pharmacy no rx needed
Online medicine home delivery: indian pharmacy delivery – indian pharmacies safe
mexican rx online: cheapest mexico drugs – mexico pharmacies prescription drugs
canadian valley pharmacy pills now even cheaper canadian pharmacy no scripts
my canadian pharmacy: pills now even cheaper – safe canadian pharmacy
india pharmacy: indian pharmacy delivery – top online pharmacy india
Online medicine home delivery Healthcare and medicines from India top 10 pharmacies in india
mexican border pharmacies shipping to usa: cheapest mexico drugs – mexican mail order pharmacies
https://canadianpharmacy24.store/# my canadian pharmacy
pharmacy in canada: Prescription Drugs from Canada – prescription drugs canada buy online
pharmacy website india Healthcare and medicines from India indian pharmacy
canadian pharmacy ratings: Certified Canadian Pharmacy – canada drug pharmacy
https://canadianpharmacy24.store/# cheap canadian pharmacy
cheapest online pharmacy india: Cheapest online pharmacy – best india pharmacy
reputable indian online pharmacy Generic Medicine India to USA top 10 pharmacies in india
mexico drug stores pharmacies: mexican pharmacy – mexican border pharmacies shipping to usa
generic amoxicillin 500mg can you buy amoxicillin over the counter medicine amoxicillin 500
https://clomidall.com/# how can i get clomid price
https://clomidall.com/# can i purchase cheap clomid without insurance
zithromax 500 where can i buy zithromax capsules zithromax capsules price
20 mg of prednisone: cheap prednisone online – prednisone 2 mg
can i order generic clomid now: buying clomid prices – can i get generic clomid pills
http://clomidall.shop/# where buy cheap clomid without prescription
how to buy clomid no prescription where can i buy cheap clomid without a prescription can you buy cheap clomid prices
https://prednisoneall.shop/# prednisone 20mg capsule
http://amoxilall.com/# amoxicillin 500 capsule
price for amoxicillin 875 mg can you purchase amoxicillin online amoxicillin 500mg capsule buy online
http://amoxilall.com/# amoxicillin cephalexin
http://clomidall.com/# get clomid
where can i buy cheap clomid without prescription can i order cheap clomid prices where buy clomid without insurance
zithromax over the counter uk: buy zithromax online australia – zithromax online australia
http://amoxilall.com/# order amoxicillin online
how to buy clomid without rx: where can i buy generic clomid pills – where buy clomid online
cost of prednisone 5mg tablets prednisone prescription drug prednisone brand name in usa
https://zithromaxall.shop/# zithromax price south africa
http://amoxilall.shop/# amoxicillin 875 125 mg tab
zithromax cost uk where to get zithromax where can i buy zithromax medicine
http://clomidall.com/# can you get generic clomid
over the counter prednisone cheap prednisone prednisone 10mg tablet price
https://clomidall.com/# can i buy cheap clomid no prescription
2.5 mg prednisone daily: purchase prednisone – prednisone cost in india
https://clomidall.shop/# how to get generic clomid for sale
cost of prednisone 10mg tablets: by prednisone w not prescription – prednisone without prescription
can you get generic clomid without rx can you get generic clomid where can i buy clomid no prescription
https://prednisoneall.shop/# over the counter prednisone cheap
generic sildenafil: Viagra generic over the counter – Viagra Tablet price
Kamagra 100mg price kamagra best price Kamagra 100mg
Cialis 20mg price in USA: Generic Tadalafil 20mg price – Generic Cialis price
Kamagra 100mg: Kamagra Iq – Kamagra tablets
viagra canada: generic ed pills – Cheapest Sildenafil online
cialis for sale cialis without a doctor prescription cheapest cialis
http://tadalafiliq.com/# Generic Tadalafil 20mg price
cheapest viagra: Buy Viagra online cheap – best price for viagra 100mg
buy kamagra online usa Kamagra Oral Jelly Price Kamagra 100mg price
Viagra Tablet price: buy viagra online – Generic Viagra online
buy Kamagra: Sildenafil Oral Jelly – super kamagra
buy Kamagra Kamagra Iq Kamagra 100mg price
Cialis over the counter: tadalafil iq – Cheap Cialis
http://sildenafiliq.xyz/# best price for viagra 100mg
Cialis 20mg price in USA: Generic Tadalafil 20mg price – Tadalafil Tablet
sildenafil oral jelly 100mg kamagra Kamagra Oral Jelly Price cheap kamagra
https://tadalafiliq.com/# п»їcialis generic
buy kamagra online usa: Kamagra Oral Jelly Price – Kamagra 100mg price
Kamagra 100mg price kamagra best price buy kamagra online usa
http://kamagraiq.shop/# super kamagra
cheap kamagra: kamagra best price – kamagra
Tadalafil Tablet: Buy Cialis online – Tadalafil price
Generic Viagra online: generic ed pills – Sildenafil Citrate Tablets 100mg
п»їcialis generic cialis for sale Cialis over the counter
https://kamagraiq.shop/# Kamagra Oral Jelly
https://sildenafiliq.xyz/# order viagra
Cheapest Sildenafil online: best price on viagra – Viagra Tablet price
Kamagra Oral Jelly: Kamagra gel – Kamagra 100mg price
https://tadalafiliq.shop/# Generic Cialis without a doctor prescription
cheap kamagra Kamagra gel Kamagra 100mg price
buy Viagra over the counter: generic ed pills – over the counter sildenafil
http://sildenafiliq.com/# viagra canada
Viagra without a doctor prescription Canada generic ed pills п»їBuy generic 100mg Viagra online
Tadalafil Tablet: Generic Tadalafil 20mg price – Generic Cialis without a doctor prescription
Kamagra 100mg price: Kamagra Oral Jelly Price – Kamagra tablets
https://tadalafiliq.shop/# Generic Tadalafil 20mg price
http://kamagraiq.shop/# cheap kamagra
Buy Tadalafil 20mg: cialis best price – Buy Tadalafil 20mg
cheapest cialis cialis best price Generic Cialis price
canadian pharmacy 365 CIPA approved pharmacies canadian pharmacy store
https://mexicanpharmgrx.shop/# buying prescription drugs in mexico
buy medicines online in india: top online pharmacy india – indianpharmacy com
buy prescription drugs from india Healthcare and medicines from India indian pharmacies safe
canadian pharmacy service: canadian pharmacy – canadianpharmacymeds com
http://mexicanpharmgrx.com/# mexican border pharmacies shipping to usa
https://mexicanpharmgrx.shop/# buying prescription drugs in mexico
canadian pharmacy world Certified Canadian pharmacies www canadianonlinepharmacy
Online medicine order indian pharmacy online shopping pharmacy india
https://canadianpharmgrx.xyz/# legitimate canadian online pharmacies
canada pharmacy reviews: Cheapest drug prices Canada – my canadian pharmacy rx
indian pharmacies safe Healthcare and medicines from India online pharmacy india
medicine in mexico pharmacies: Mexico drugstore – mexican online pharmacies prescription drugs
http://indianpharmgrx.shop/# top 10 online pharmacy in india
best online pharmacy india indian pharmacy online shopping pharmacy india
recommended canadian pharmacies CIPA approved pharmacies canadian pharmacy 365
http://mexicanpharmgrx.com/# reputable mexican pharmacies online
http://mexicanpharmgrx.com/# reputable mexican pharmacies online
best online pharmacies in mexico: Pills from Mexican Pharmacy – п»їbest mexican online pharmacies
mail order pharmacy india indian pharmacy reputable indian online pharmacy
india online pharmacy: Generic Medicine India to USA – buy medicines online in india
http://indianpharmgrx.shop/# Online medicine order
mexican border pharmacies shipping to usa Pills from Mexican Pharmacy mexican drugstore online
https://mexicanpharmgrx.shop/# mexico drug stores pharmacies
http://indianpharmgrx.com/# reputable indian pharmacies
best india pharmacy indian pharmacy indian pharmacy paypal
https://canadianpharmgrx.com/# canadian pharmacy oxycodone
aromatase inhibitor tamoxifen: tamoxifen lawsuit – tamoxifen moa
buy cytotec online Cytotec 200mcg price cytotec online
how to buy doxycycline online: doxycycline – buy cheap doxycycline
where can i buy diflucan over the counter diflucan 200mg tab buy diflucan yeast infection
doxycycline 50mg: buy doxycycline – doxycycline without a prescription
https://doxycyclinest.pro/# doxycycline hyc 100mg
tamoxifen benefits: who should take tamoxifen – tamoxifen and antidepressants
doxycycline tablets doxy buy doxycycline for dogs
buy cytotec over the counter: cytotec buy online usa – cytotec pills online
buy doxycycline order doxycycline online buy doxycycline online
cipro for sale: cipro 500mg best prices – buy ciprofloxacin
diflucan usa: diflucan online purchase – diflucan 50 mg capsule
nolvadex steroids: where to get nolvadex – tamoxifen postmenopausal
order cytotec online buy cytotec pills online cheap cytotec abortion pill
http://ciprofloxacin.guru/# ciprofloxacin mail online
purchase doxycycline online: buy doxycycline monohydrate – online doxycycline
purchase cytotec buy cytotec in usa cytotec pills buy online
buy cytotec over the counter: buy cytotec in usa – cytotec pills online
cytotec abortion pill: buy cytotec in usa – Cytotec 200mcg price
doxycycline 100mg capsules: doxycycline 150 mg – buy doxycycline online
effexor and tamoxifen: tamoxifen and depression – dcis tamoxifen
https://diflucan.icu/# where can i get diflucan
buy cytotec over the counter buy cytotec pills online cheap Abortion pills online
doxycycline online: doxycycline 100mg capsules – buy doxycycline monohydrate
what happens when you stop taking tamoxifen: alternatives to tamoxifen – tamoxifen and uterine thickening
diflucan 200 mg daily diflucan cost in india can you buy diflucan over the counter
doxycycline 200 mg: doxycycline 100mg online – where to get doxycycline
odering doxycycline: doxycycline 100 mg – doxycycline online
order doxycycline doxy doxycycline 100mg price
diflucan 150 mg tablet price in india: diflucan rx online – where to buy diflucan pills
where can i buy amoxicillin without prec: buy amoxicillin without prescription – prescription for amoxicillin
clomid cost where can i buy cheap clomid price can i order generic clomid pill
can you buy amoxicillin over the counter canada: amoxicillin 500 coupon – amoxicillin 500mg cost
amoxicillin 875 125 mg tab order amoxicillin 500mg amoxicillin without rx
http://prednisonea.store/# prednisone in mexico
prednisone for sale online: how to purchase prednisone online – prednisone buy without prescription
https://stromectola.top/# ivermectin 400 mg brands
amoxicillin 500mg tablets price in india: amoxicillin discount coupon – amoxicillin 500 mg tablets
prednisone: generic prednisone for sale – prednisone buy
zithromax 600 mg tablets zithromax for sale 500 mg can i buy zithromax over the counter
http://stromectola.top/# ivermectin pills
zithromax pill: average cost of generic zithromax – zithromax 250
stromectol price usa stromectol cvs ivermectin 400 mg brands
buy amoxicillin online uk: amoxicillin 500mg capsules price – purchase amoxicillin 500 mg
https://clomida.pro/# how can i get clomid without a prescription
prednisone pharmacy: prednisone without prescription.net – medicine prednisone 5mg
amoxicillin 500mg capsule buy online amoxicillin 500mg without prescription antibiotic amoxicillin
https://stromectola.top/# order minocycline 50mg
https://clomida.pro/# can i get cheap clomid online
stromectol where to buy: stromectol pill – stromectol 3 mg tablet
prescription drugs from canada: canada pharmacy not requiring prescription – offshore pharmacy no prescription
http://medicationnoprescription.pro/# how to get a prescription in canada
non prescription online pharmacy india ordering prescription drugs from canada online pharmacy canada no prescription
https://medicationnoprescription.pro/# online pharmacy without a prescription
erectile dysfunction online prescription: low cost ed medication – ed medications cost
canada prescription online no prescription drugs canada prescription drugs online
http://medicationnoprescription.pro/# canadian pharmacy without prescription
https://edpill.top/# online ed drugs
drugstore com online pharmacy prescription drugs: international pharmacy no prescription – buying prescription drugs from canada
prescription meds from canada: online pharmacy without prescription – buying prescription drugs online without a prescription
http://edpill.top/# ed online treatment
get ed meds online online ed meds erectile dysfunction meds online
http://edpill.top/# what is the cheapest ed medication
online meds without prescription: canadian prescriptions in usa – no prescription pharmacy
order ed pills online affordable ed medication how to get ed meds online
https://onlinepharmacyworld.shop/# cheap pharmacy no prescription
online meds no prescription: buy prescription drugs on line – cheap prescription medication online
http://edpill.top/# buying ed pills online
https://edpill.top/# online ed prescription
erectile dysfunction pills for sale cheapest ed online ed doctor online
http://casinvietnam.com/# web c? b?c online uy tin
casino online uy tín: dánh bài tr?c tuy?n – choi casino tr?c tuy?n trên di?n tho?i
Thanks for the good critique. Me and my cousin were just preparing to do a little research on this. We got a book from our area library but I think I’ve learned better from this post. I’m very glad to see such wonderful information being shared freely out there..
http://casinvietnam.shop/# choi casino tr?c tuy?n tren di?n tho?i
casino online uy tin game c? b?c online uy tin casino tr?c tuy?n uy tin
I would like to show some thanks to you for rescuing me from such a instance. Just after searching through the world-wide-web and coming across recommendations which are not helpful, I was thinking my life was gone. Being alive devoid of the answers to the issues you have fixed as a result of this review is a serious case, as well as ones which could have in a wrong way damaged my career if I had not discovered your web blog. Your main mastery and kindness in touching almost everything was helpful. I’m not sure what I would’ve done if I hadn’t come upon such a stuff like this. It’s possible to at this time look ahead to my future. Thanks for your time so much for the expert and result oriented guide. I will not think twice to recommend your web site to anybody who will need recommendations about this matter.
clomid tablets over the counter
https://casinvietnam.com/# choi casino tr?c tuy?n tren di?n tho?i
https://casinvietnam.com/# choi casino tr?c tuy?n tren di?n tho?i
http://casinvietnam.shop/# casino online uy tin
https://casinvietnam.shop/# casino online uy tin
http://casinvietnam.com/# casino tr?c tuy?n vi?t nam
casino tr?c tuy?n choi casino tr?c tuy?n tren di?n tho?i casino tr?c tuy?n
http://casinvietnam.shop/# web c? b?c online uy tin
danh bai tr?c tuy?n casino tr?c tuy?n vi?t nam casino tr?c tuy?n uy tin
http://casinvietnam.com/# choi casino tr?c tuy?n tren di?n tho?i
casino tr?c tuy?n uy tin choi casino tr?c tuy?n tren di?n tho?i casino tr?c tuy?n
https://casinvietnam.shop/# game c? b?c online uy tin
web c? b?c online uy tin danh bai tr?c tuy?n casino online uy tin
https://casinvietnam.shop/# casino tr?c tuy?n
casino tr?c tuy?n vi?t nam casino online uy tin casino tr?c tuy?n vi?t nam
https://casinvietnam.shop/# casino tr?c tuy?n uy tin
choi casino tr?c tuy?n tren di?n tho?i choi casino tr?c tuy?n tren di?n tho?i game c? b?c online uy tin
https://casinvietnam.com/# casino tr?c tuy?n vi?t nam
https://casinvietnam.shop/# choi casino tr?c tuy?n tren di?n tho?i
web c? b?c online uy tin casino online uy tin casino tr?c tuy?n vi?t nam
mexico drug stores pharmacies medicine in mexico pharmacies mexican border pharmacies shipping to usa
http://indiaph24.store/# online pharmacy india
best online pharmacies in mexico mexico pharmacy medicine in mexico pharmacies
canadian pharmacy checker Prescription Drugs from Canada cross border pharmacy canada
http://indiaph24.store/# Online medicine home delivery
top online pharmacy india buy medicines from India top 10 online pharmacy in india
best india pharmacy buy medicines from India india pharmacy mail order
http://canadaph24.pro/# best mail order pharmacy canada
There are extremely lots of details that adheres to that take into consideration. That is a wonderful indicate retrieve. I provide you with the thoughts above as general inspiration but clearly there are questions such as one you raise up where the biggest thing will be employed in honest good faith. I don?t know if guidelines have emerged about items like that, but I am certain that the job is clearly referred to as a good game. Both kids have the impact of just a moment’s pleasure, for the rest of their lives.
mexico pharmacies prescription drugs mexican drugstore online mexican rx online
mexico drug stores pharmacies Online Pharmacies in Mexico pharmacies in mexico that ship to usa
http://mexicoph24.life/# buying prescription drugs in mexico
online canadian pharmacy reviews canadian pharmacies canadian pharmacy king
Online medicine order indian pharmacy Online medicine order
http://canadaph24.pro/# prescription drugs canada buy online
top 10 pharmacies in india buy medicines from India Online medicine home delivery
buy prescription drugs from india Generic Medicine India to USA top online pharmacy india
http://mexicoph24.life/# mexican pharmaceuticals online
top 10 pharmacies in india Generic Medicine India to USA reputable indian pharmacies
Greetings! I’ve been reading your website for a long time now and finally got the bravery to go ahead and give you a shout out from Dallas Tx! Just wanted to say keep up the good job!
canadian neighbor pharmacy Certified Canadian Pharmacies cheap canadian pharmacy
https://indiaph24.store/# online pharmacy india
best online pharmacy india Generic Medicine India to USA top 10 pharmacies in india
my canadian pharmacy Licensed Canadian Pharmacy canadian pharmacy
https://canadaph24.pro/# canadian pharmacy 1 internet online drugstore
Online medicine order Cheapest online pharmacy best online pharmacy india
canadian pharmacy ltd Certified Canadian Pharmacies best canadian pharmacy online
Hi there! Good post! Please do tell us when I could see a follow up!
https://mexicoph24.life/# mexican online pharmacies prescription drugs
mail order pharmacy india buy medicines from India top 10 pharmacies in india
Found this on Yahoo and I’m glad I did. Well written article.
canadian pharmacy meds review Certified Canadian Pharmacies best canadian pharmacy to buy from
mexican mail order pharmacies Mexican Pharmacy Online buying from online mexican pharmacy
best online pharmacies in mexico Online Pharmacies in Mexico п»їbest mexican online pharmacies
http://canadaph24.pro/# pharmacies in canada that ship to the us
reputable indian online pharmacy Generic Medicine India to USA best india pharmacy
canadian pharmacy review Prescription Drugs from Canada canadian pharmacy meds
canadian pharmacy sarasota Large Selection of Medications from Canada legit canadian pharmacy
https://canadaph24.pro/# safe reliable canadian pharmacy
cheapest online pharmacy india Online medicine home delivery top online pharmacy india
There are also many conditions with symptoms similar to appendicitis. But because appendicitis can become serious in a short amount of time, call your
top 10 pharmacies in india http://indiaph24.store/# buy medicines online in india
mail order pharmacy india
top 10 pharmacies in india Generic Medicine India to USA indian pharmacy online
Online medicine home delivery Cheapest online pharmacy best india pharmacy
https://mexicoph24.life/# pharmacies in mexico that ship to usa
mexico pharmacies prescription drugs Mexican Pharmacy Online pharmacies in mexico that ship to usa
indian pharmacy online indian pharmacy fast delivery buy prescription drugs from india
buy prescription drugs from india Cheapest online pharmacy online pharmacy india
https://mexicoph24.life/# buying from online mexican pharmacy
vipps canadian pharmacy onlinecanadianpharmacy 24 canadian pharmacy store
Can I use my insurance for fertility medications clomiphene Citrate men over 50?
india pharmacy mail order indian pharmacy Online medicine home delivery
http://canadaph24.pro/# canada pharmacy world
certified canadian pharmacy Certified Canadian Pharmacies canadian pharmacy 365
buying prescription drugs in mexico mexican pharmacy mexican pharmacy
http://indiaph24.store/# cheapest online pharmacy india
best india pharmacy Cheapest online pharmacy world pharmacy india
п»їbest mexican online pharmacies Online Pharmacies in Mexico reputable mexican pharmacies online
legit canadian pharmacy online canadian pharmacies pharmacies in canada that ship to the us
http://canadaph24.pro/# reliable canadian pharmacy reviews
mexico pharmacy mexico pharmacy mexico drug stores pharmacies
reputable mexican pharmacies online mexican pharmacy pharmacies in mexico that ship to usa
Spot lets start on this write-up, I honestly think this website needs much more consideration. I’ll oftimes be once more you just read a lot more, thank you for that info.
http://mexicoph24.life/# mexican pharmaceuticals online
https://indiaph24.store/# india pharmacy
https://indiaph24.store/# top 10 pharmacies in india
pharmacies in mexico that ship to usa: cheapest mexico drugs – mexico pharmacies prescription drugs
I rather encourage you to learn about the flexibility of forms, fields and widgets and how it’s used in the automatic Admin interface.
medication canadian pharmacy Certified Canadian Pharmacies northwest canadian pharmacy
canadian neighbor pharmacy Licensed Canadian Pharmacy canadian pharmacy no scripts
https://mexicoph24.life/# medication from mexico pharmacy
п»їbest mexican online pharmacies: mexican pharmacy – purple pharmacy mexico price list
http://canadaph24.pro/# canadian pharmacy near me
reputable mexican pharmacies online Mexican Pharmacy Online mexican pharmaceuticals online
Great post. The tips and the ideas given in the post seems to be very much informative and useful
http://ciprofloxacin.tech/# ciprofloxacin 500 mg tablet price
tamoxifen mechanism of action: lexapro and tamoxifen – tamoxifen depression
tamoxifen therapy pct nolvadex tamoxifen alternatives
get propecia no prescription cheap propecia without rx cost of propecia price
Howdy! Do you use Twitter? I’d like to follow you if that would be okay. I’m absolutely enjoying your blog and look forward to new updates.
http://nolvadex.life/# tamoxifenworld
cost of cheap propecia without insurance: order cheap propecia tablets – cost generic propecia pill
lisinopril 5 mg tablet lisinopril generic cost lisinopril tab 5 mg price
https://nolvadex.life/# tamoxifen chemo
buy misoprostol over the counter: order cytotec online – Abortion pills online
cipro pharmacy buy cipro online without prescription buy cipro online without prescription
http://cytotec.club/# buy cytotec over the counter
http://lisinopril.network/# lisinopril 40 mg india
Abortion pills online: buy cytotec over the counter – order cytotec online
https://finasteride.store/# buy generic propecia without a prescription
Misoprostol 200 mg buy online cytotec abortion pill cytotec buy online usa
https://cytotec.club/# Cytotec 200mcg price
lisinopril tablets india: lisinopril 10 mg prices – lisinopril 40mg prescription cost
i love action movies and my idol is none other than Gerard Butler. this guy really rocks“
buy propecia price buy propecia prices propecia brand name
prinivil generic: lisinopril 1.25 – lisinopril 80 mg
generic propecia without dr prescription order cheap propecia without insurance buying cheap propecia no prescription
https://ciprofloxacin.tech/# buy generic ciprofloxacin
tamoxifen breast cancer prevention: tamoxifen and bone density – tamoxifenworld
I am glad to be one of the visitants on this outstanding website (:, appreciate it for posting .
https://nolvadex.life/# tamoxifen hip pain
prescription drug prices lisinopril lisinopril 420 1g average cost of lisinopril
cytotec pills buy online Abortion pills online п»їcytotec pills online
https://cytotec.club/# buy cytotec pills
ciprofloxacin 500mg buy online: buy cipro online canada – cipro pharmacy
I have been reading out a few of your stories and it’s pretty nice stuff. I will make sure to bookmark your site
http://cytotec.club/# Cytotec 200mcg price
cipro online no prescription in the usa: cipro – cipro online no prescription in the usa
tamoxifen alternatives nolvadex estrogen blocker tamoxifen and osteoporosis
buy cytotec over the counter buy cytotec pills Abortion pills online
https://ciprofloxacin.tech/# cipro
buy cytotec over the counter: Abortion pills online – buy cytotec pills
http://cytotec.club/# buy cytotec
buy ciprofloxacin cipro online no prescription in the usa cipro for sale
cytotec buy online usa: cytotec abortion pill – order cytotec online
cytotec buy online usa buy cytotec online cytotec online
https://finasteride.store/# get generic propecia without insurance
https://ciprofloxacin.tech/# where can i buy cipro online
tamoxifenworld: tamoxifen men – tamoxifen
cost propecia cost of cheap propecia now buy generic propecia without prescription
http://ciprofloxacin.tech/# buy cipro
home propecia price cost of generic propecia
https://ciprofloxacin.tech/# buy cipro cheap
purchase cipro: cipro pharmacy – ciprofloxacin generic
http://finasteride.store/# buying propecia pill
where can i buy cipro online: purchase cipro – ciprofloxacin 500mg buy online
antibiotics cipro ciprofloxacin 500 mg tablet price cipro pharmacy
ciprofloxacin mail online buy cipro online without prescription cipro pharmacy
http://nolvadex.life/# nolvadex d
ciprofloxacin 500mg buy online: ciprofloxacin generic – buy cipro
http://ciprofloxacin.tech/# cipro ciprofloxacin
otc lisinopril: how much is lisinopril 5 mg – lisinopril online pharmacy
tamoxifen hair loss tamoxifen mechanism of action tamoxifen and bone density
buy cytotec pills online cheap Cytotec 200mcg price buy cytotec online fast delivery
https://finasteride.store/# cost of generic propecia tablets
https://cytotec.club/# п»їcytotec pills online
Abortion pills online: cytotec pills online – Abortion pills online
http://cytotec.club/# cytotec online
order cytotec online buy cytotec pills cytotec buy online usa
buy cheap propecia without insurance get generic propecia without a prescription buying propecia without rx
can i order lisinopril online: zestril 20 mg cost – lisinopril 40 mg best price
Are there pills that should be taken with specific religious practices kamagra online?
https://lisinopril.network/# lisinopril price
cytotec pills buy online: Misoprostol 200 mg buy online – cytotec abortion pill
cytotec abortion pill cytotec buy online usa buy cytotec in usa
cost of propecia without dr prescription cost of propecia pill buy propecia
http://nolvadex.life/# tamoxifen dosage
order cheap propecia no prescription: cost of cheap propecia online – order cheap propecia tablets
https://nolvadex.life/# tamoxifen and osteoporosis
https://nolvadex.life/# tamoxifen citrate pct
buy cytotec online fast delivery: cytotec buy online usa – purchase cytotec
cost of brand name lisinopril prinivil 2.5 mg buy zestril
tamoxifen therapy tamoxifen therapy nolvadex for sale amazon
https://ciprofloxacin.tech/# cipro for sale
https://cytotec.club/# buy cytotec online
https://levitrav.store/# Levitra online USA fast
cheapest cenforce: Buy Cenforce 100mg Online – Purchase Cenforce Online
https://cialist.pro/# Buy Tadalafil 20mg
levitra reviews
Levitra tablet price: Vardenafil online prescription – Vardenafil price
Cheap generic Viagra Buy Viagra online cheap buy Viagra online
Order Viagra 50 mg online: Cheapest place to buy Viagra – buy Viagra online
buy kamagra online usa kamagra pills Kamagra 100mg
https://levitrav.store/# Levitra 20 mg for sale
Cheap generic Viagra: Buy Viagra online – sildenafil online
https://cialist.pro/# Buy Cialis online
https://levitrav.store/# Buy Vardenafil online
Kamagra 100mg: Kamagra Oral Jelly – Kamagra 100mg
buy cenforce Purchase Cenforce Online cenforce for sale
Levitra 10 mg best price Buy Vardenafil 20mg Buy Vardenafil 20mg
https://viagras.online/# best price for viagra 100mg
cheapest cenforce: cenforce.pro – Buy Cenforce 100mg Online
sildenafil over the counter Cheapest place to buy Viagra п»їBuy generic 100mg Viagra online
Generic Viagra online viagras.online cheap viagra
http://cenforce.pro/# cheapest cenforce
Vardenafil online prescription: Levitra generic price – buy Levitra over the counter
Generic Cialis without a doctor prescription: buy cialis online – Buy Tadalafil 20mg
generic sildenafil: Buy Viagra online – Generic Viagra online
https://levitrav.store/# Buy Levitra 20mg online
cenforce for sale: Cenforce 150 mg online – cheapest cenforce
Kamagra 100mg price buy kamagra online buy Kamagra
order cenforce: cheapest cenforce – cenforce for sale
A formidable share, I just given this onto a colleague who was doing just a little analysis on this. And he in reality purchased me breakfast as a result of I discovered it for him.. smile. So let me reword that: Thnx for the treat! But yeah Thnkx for spending the time to debate this, I really feel strongly about it and love reading extra on this topic. If potential, as you turn out to be expertise, would you mind updating your weblog with more particulars? It’s extremely useful for me. Big thumb up for this weblog submit!
https://cenforce.pro/# Purchase Cenforce Online
buy cialis pill: buy cialis overseas – Buy Tadalafil 10mg
cheap kamagra buy kamagra online sildenafil oral jelly 100mg kamagra
https://levitrav.store/# Vardenafil buy online
https://kamagra.win/# Kamagra 100mg price
Buy Cialis online buy cialis online п»їcialis generic
https://viagras.online/# viagra canada
https://levitrav.store/# Vardenafil price
https://kamagra.win/# Kamagra 100mg price
https://levitrav.store/# Buy Levitra 20mg online
Its wonderful as your other articles : D, regards for posting .
order viagra Buy Viagra online cheap п»їBuy generic 100mg Viagra online
cialis for sale: buy cialis overseas – Generic Tadalafil 20mg price
sildenafil oral jelly 100mg kamagra п»їkamagra Kamagra 100mg price
http://viagras.online/# Sildenafil 100mg price
Kamagra tablets: kamagra oral jelly – Kamagra 100mg price
Cheap Levitra online: buy Levitra over the counter – Levitra 10 mg best price
Tadalafil Tablet: cialist.pro – Generic Tadalafil 20mg price
http://kamagra.win/# Kamagra 100mg price
Viagra without a doctor prescription Canada Buy generic 100mg Viagra online Viagra generic over the counter
https://pharmindia.online/# indianpharmacy com
india pharmacy mail order: best online pharmacy india – Online medicine order
cheap canadian pharmacy online: canadian pharmacy uk delivery – safe online pharmacies in canada
mail order pharmacy india indian pharmacy online Online medicine home delivery
medication from mexico pharmacy: mexican rx online – mexican online pharmacies prescription drugs
best online pharmacies in mexico best online pharmacies in mexico mexican online pharmacies prescription drugs
Pretty! This was a really wonderful post. Thank you for your provided information.
https://pharmcanada.shop/# canada drug pharmacy
medication from mexico pharmacy: mexican drugstore online – buying from online mexican pharmacy
https://pharmworld.store/# rxpharmacycoupons
http://pharmnoprescription.icu/# canada mail order prescriptions
http://pharmcanada.shop/# canadian pharmacy world
cheapest pharmacy for prescriptions without insurance: canadian pharmacy coupon code – no prescription pharmacy paypal
purple pharmacy mexico price list mexico pharmacy buying prescription drugs in mexico
india pharmacy reputable indian online pharmacy buy prescription drugs from india
I like this post, enjoyed this one thanks for putting up.
my canadian pharmacy review: safe reliable canadian pharmacy – canadian pharmacy king
https://pharmworld.store/# canadian pharmacy discount coupon
my canadian pharmacy: canadian pharmacy uk delivery – pharmacy in canada
reputable indian online pharmacy: best india pharmacy – buy prescription drugs from india
no prescription required pharmacy: online pharmacy – online pharmacy non prescription drugs
canadian pharmacy phone number canadian pharmacy cheap canadian mail order pharmacy
online pharmacy no prescription international pharmacy no prescription canadian pharmacy no prescription needed
mexican rx online: reputable mexican pharmacies online – medicine in mexico pharmacies
https://pharmmexico.online/# mexican pharmacy
Great post, thanks for the awesome blog post. I’m having troubles subscribing to your blogs feed. Thought I’d let you know
I got what you intend, appreciate it for posting .Woh I am lucky to find this website through google.
best online pharmacy india: best online pharmacy india – reputable indian pharmacies
http://pharmmexico.online/# mexican pharmacy
canadian pharmacy 365 pharmacy com canada pharmacy canadian superstore
best online pharmacy india buy medicines online in india п»їlegitimate online pharmacies india
buy prescription drugs from india: Online medicine home delivery – india pharmacy
http://pharmcanada.shop/# canadian pharmacy ratings
online shopping pharmacy india: india pharmacy – indian pharmacy online
http://pharmnoprescription.icu/# order prescription from canada
canadian pharmacy coupon code canadian pharmacy world coupon code legit non prescription pharmacies
legitimate canadian pharmacy online canadian online pharmacy canada drugstore pharmacy rx
canadian family pharmacy: legitimate canadian pharmacy – canada pharmacy online
Excellent commentary! Thought about was pleased with their scanning. I hope to enjoy a book way more of you. It looks you’ll have very good understanding and so dream. Now i am particularly contented in this guidance.
https://pharmindia.online/# top 10 online pharmacy in india
buy prescription drugs from canada cheap: online canadian pharmacy – canada pharmacy world
uk pharmacy no prescription: online pharmacy – canadian pharmacy coupon code
mexican mail order pharmacies: buying prescription drugs in mexico – buying prescription drugs in mexico online
http://pharmcanada.shop/# buy drugs from canada
legitimate canadian pharmacy: canadian pharmacy oxycodone – canadian pharmacy meds reviews
canada pharmacy online canadian drug best canadian online pharmacy
canadian pharmacy coupon online pharmacy online pharmacy discount code
reliable canadian pharmacy: canada drugs online reviews – canadianpharmacymeds
http://pharmindia.online/# india pharmacy
indian pharmacies safe: Online medicine home delivery – cheapest online pharmacy india
canada pharmacy coupon: pharm world store – pharmacy coupons
http://pharmnoprescription.icu/# meds online no prescription
azithromycin amoxicillin generic amoxicillin how to buy amoxycillin
can we buy amoxcillin 500mg on ebay without prescription: amoxicillin for sale – where can i buy amoxicillin over the counter
buy zithromax without presc buy zithromax without prescription online buy cheap zithromax online
zithromax 250 mg: how to buy zithromax online – can you buy zithromax over the counter in australia
where can i get prednisone over the counter: cheapest prednisone no prescription – prednisone sale
Yeah bookmaking this wasn’t a speculative conclusion great post! .
http://zithromaxa.store/# zithromax prescription
neurontin 300 mg cost: neurontin 204 – gabapentin online
where to get zithromax: where can i get zithromax over the counter – cheap zithromax pills
https://doxycyclinea.online/# doxycycline monohydrate
where to buy amoxicillin 500mg prescription for amoxicillin buy amoxicillin 500mg capsules uk
amoxicillin 500 mg online amoxicillin without rx can you buy amoxicillin over the counter in canada
prednisone 5 mg tablet cost: prednisone in mexico – no prescription online prednisone
average cost of prednisone: prednisone 5 mg brand name – prednisone without prescription
https://gabapentinneurontin.pro/# how to get neurontin
How can I verify the online pharmacy’s policy on environmental sustainability vidalista 20?
neurontin 800 pill: neurontin cost – neurontin for sale online
amoxicillin script: amoxicillin 500mg cost – amoxicillin buy online canada
prednisone 10 tablet: prednisone 20mg tablets where to buy – prednisone 20mg capsule
doxycycline: odering doxycycline – doxycycline without prescription
where can i buy amoxicillin over the counter: generic amoxicillin online – amoxicillin 500 mg tablets
buy doxycycline cheap: doxy 200 – how to buy doxycycline online
prednisone drug costs: how much is prednisone 5mg – ordering prednisone
zithromax pill: zithromax online no prescription – zithromax generic price
5 mg prednisone tablets prednisone oral buy prednisone online india
prednisone 10 buy prednisone from india fast shipping prednisone
order prednisone online no prescription: prednisone 20mg – prednisone 5093
https://zithromaxa.store/# can you buy zithromax online
prednisone brand name in usa: prednisone 5 mg tablet – prednisone 10mg cost
amoxicillin 500mg tablets price in india: cheap amoxicillin 500mg – can you buy amoxicillin over the counter in canada
price for amoxicillin 875 mg: amoxicillin 500 mg where to buy – amoxicillin 250 mg
doxycycline hyc 100mg price of doxycycline doxycycline 100mg
https://prednisoned.online/# price of prednisone tablets
buy generic neurontin generic gabapentin neurontin for sale
buy neurontin 100 mg: neurontin 300 mg buy – neurontin discount
https://zithromaxa.store/# buy azithromycin zithromax
zithromax antibiotic: buy zithromax online australia – zithromax prescription in canada
zithromax 250 mg australia: zithromax coupon – can you buy zithromax over the counter in australia
purchase neurontin canada neurontin uk neurontin capsule 600mg
doxycycline 100mg capsules doxycycline 100 mg online doxycycline
doxycycline prices: where to purchase doxycycline – doxycycline 500mg
prednisone otc uk: prednisone 10 – over the counter prednisone pills
https://doxycyclinea.online/# online doxycycline
buy amoxicillin without prescription: buy amoxicillin online with paypal – amoxicillin 500
prednisone pills cost prednisone 60 mg daily prednisone generic brand name
http://zithromaxa.store/# zithromax cost canada
Well, this Tuesday I read through a couple of your posts and this is probably one of your better ones. ..
buy zithromax 500mg online: zithromax generic price – order zithromax without prescription
prednisone 20mg by mail order: prednisone canada pharmacy – buy prednisone online no prescription
https://gabapentinneurontin.pro/# neurontin 1800 mg
zithromax price canada: zithromax azithromycin – zithromax 500 without prescription
prednisone without prescription.net prednisone purchase canada prednisone 2.5 mg cost
200 mg doxycycline doxycycline hyclate doxycycline mono
https://gabapentinneurontin.pro/# neurontin drug
how to buy doxycycline online: buy doxycycline hyclate 100mg without a rx – buy doxycycline without prescription
doxycycline pills: 100mg doxycycline – buy doxycycline online without prescription
https://zithromaxa.store/# zithromax online no prescription
can you buy amoxicillin uk: amoxicillin no prescription – amoxicillin 500mg cost
buy cheap neurontin online neurontin canada neurontin 400
zithromax 500 price purchase zithromax online zithromax 250 mg australia
https://prednisoned.online/# canada buy prednisone online
60 mg prednisone daily: order prednisone 10 mg tablet – prednisone
http://gabapentinneurontin.pro/# neurontin 800 mg
order amoxicillin uk: buy amoxicillin 500mg canada – amoxicillin 825 mg
cost of generic zithromax where to get zithromax over the counter zithromax pill
buying amoxicillin online antibiotic amoxicillin prescription for amoxicillin
generic zithromax 500mg india: zithromax 500mg over the counter – zithromax buy
zithromax 600 mg tablets: zithromax z-pak – zithromax 600 mg tablets
purchase neurontin online: buy generic neurontin – neurontin 3
What your declaring is absolutely true. I know that everyone need to say the similar factor, but I just feel that you set it in a way that everybody can comprehend. I also love the images you place in right here. They suit so well with what youre attempting to say. Im sure youll attain so quite a few folks with what youve received to say.
how to get zithromax online: zithromax buy online – cost of generic zithromax
https://gabapentinneurontin.pro/# neurontin 400 mg capsules
amoxicillin online canada amoxicillin capsule 500mg price amoxicillin pharmacy price
doxy doxycycline 100mg online doxycycline 500mg
prednisone purchase online: online prednisone 5mg – prednisone 20mg price in india
can you buy prednisone over the counter in canada: 20mg prednisone – where to buy prednisone 20mg no prescription
https://doxycyclinea.online/# price of doxycycline
neurontin canada online: neurontin 400 mg capsules – how much is generic neurontin
http://doxycyclinea.online/# doxycycline
doxycycline 50mg: doxycycline order online – generic for doxycycline
where can i get zithromax over the counter zithromax online paypal azithromycin zithromax
where can i buy amoxocillin buy amoxil amoxicillin over counter
canada where to buy neurontin: neurontin 300 mg price – brand name neurontin
http://prednisoned.online/# can you buy prednisone without a prescription
odering doxycycline: order doxycycline – buy doxycycline 100mg
https://amoxila.pro/# buy amoxicillin online mexico
zithromax z-pak price without insurance buy cheap zithromax online zithromax 500mg
buy zithromax online cheap: zithromax antibiotic – how to buy zithromax online
buy doxycycline 100mg: doxycycline 500mg – doxycycline vibramycin
buy neurontin neurontin singapore neurontin 3
amoxicillin over the counter in canada: amoxicillin without prescription – how to buy amoxicillin online
http://gabapentinneurontin.pro/# neurontin mexico
neurontin 400 mg cost: neurontin 300mg – neurontin 300 mg price in india
https://amoxila.pro/# amoxicillin 500 mg capsule
prednisone for sale in canada: prednisone canada – prednisone over the counter uk
medicine amoxicillin 500mg amoxicillin price without insurance amoxicillin 500mg capsules uk
doxy 200 doxycycline pills generic doxycycline
buy doxycycline 100mg: doxycycline hyclate 100 mg cap – doxycycline hyclate
http://doxycyclinea.online/# buy doxycycline online
neurontin brand name 800mg best price: neurontin sale – neurontin generic
https://amoxila.pro/# cheap amoxicillin 500mg
neurontin generic brand neurontin 900 mg neurontin 300 mg price in india
cost of generic zithromax generic zithromax azithromycin zithromax prescription in canada
neurontin 100 mg caps: neurontin canada – neurontin cost australia
amoxicillin without prescription: amoxicillin 500mg capsules – where to buy amoxicillin over the counter
zithromax drug: zithromax online usa no prescription – buy zithromax 1000mg online
http://amoxila.pro/# amoxicillin 500mg capsules price
can i buy zithromax over the counter in canada: cost of generic zithromax – zithromax prescription
neurontin tablets 100mg neurontin 800 mg tablets neurontin 600 mg cost
http://gabapentinneurontin.pro/# neurontin 150 mg
prednisone pills 10 mg can you buy prednisone over the counter in mexico prednisone 5mg price
buy amoxicillin canada: ampicillin amoxicillin – amoxicillin 200 mg tablet
where to get doxycycline: buy doxycycline without prescription uk – buy doxycycline online without prescription
http://zithromaxa.store/# zithromax antibiotic without prescription
zithromax prescription online: can you buy zithromax over the counter – where can i buy zithromax uk
Can I take my pills with fortified sports drinks advair diskus cost?
generic amoxicillin medicine amoxicillin 500 buy amoxicillin
doxycycline generic: doxycycline 100mg online – buy doxycycline monohydrate
cheap doxycycline online buy doxycycline online buy doxycycline
https://prednisoned.online/# average cost of prednisone 20 mg
neurontin 100 mg caps: neurontin discount – 300 mg neurontin
zithromax 500 without prescription: generic zithromax medicine – buy zithromax without presc
http://mexicanpharmacy1st.com/# mexico drug stores pharmacies
mexico pharmacies prescription drugs: mexican online pharmacies prescription drugs – mexican online pharmacies prescription drugs
mexican rx online: buying from online mexican pharmacy – reputable mexican pharmacies online
buying from online mexican pharmacy: mexican online pharmacies prescription drugs – medication from mexico pharmacy
mexico drug stores pharmacies: medicine in mexico pharmacies – mexico drug stores pharmacies
reputable mexican pharmacies online mexican border pharmacies shipping to usa mexican online pharmacies prescription drugs
buying prescription drugs in mexico buying prescription drugs in mexico online medicine in mexico pharmacies
https://mexicanpharmacy1st.shop/# mexican mail order pharmacies
mexican online pharmacies prescription drugs: medication from mexico pharmacy – pharmacies in mexico that ship to usa
mexican drugstore online: п»їbest mexican online pharmacies – purple pharmacy mexico price list
http://mexicanpharmacy1st.com/# mexican rx online
mexico pharmacy: best online pharmacies in mexico – mexico pharmacies prescription drugs
When I originally commented I clicked the -Notify me when new comments are added- checkbox and now each time a comment is added I get four emails with the same comment. Is there any way you can remove me from that service? Thanks!
https://mexicanpharmacy1st.com/# buying prescription drugs in mexico online
Nice post. I understand some thing more challenging on distinct blogs everyday. It will always be stimulating to read content off their writers and rehearse a specific thing there. I’d would prefer to apply certain while using content in this little blog whether you do not mind. Natually I’ll offer you a link with your internet weblog. Many thanks sharing.
http://mexicanpharmacy1st.com/# mexican mail order pharmacies
mexican border pharmacies shipping to usa: pharmacies in mexico that ship to usa – mexican online pharmacies prescription drugs
mexico pharmacies prescription drugs: buying from online mexican pharmacy – pharmacies in mexico that ship to usa
п»їbest mexican online pharmacies: medication from mexico pharmacy – mexico pharmacies prescription drugs
reputable mexican pharmacies online п»їbest mexican online pharmacies mexico pharmacy
https://mexicanpharmacy1st.shop/# mexican mail order pharmacies
medicine in mexico pharmacies: best mexican online pharmacies – pharmacies in mexico that ship to usa
mexican drugstore online: buying from online mexican pharmacy – mexico drug stores pharmacies
https://mexicanpharmacy1st.com/# mexico pharmacies prescription drugs
best online pharmacies in mexico mexico drug stores pharmacies mexico pharmacy
mexico drug stores pharmacies: medicine in mexico pharmacies – mexico pharmacy
https://mexicanpharmacy1st.com/# mexico drug stores pharmacies
https://mexicanpharmacy1st.shop/# medication from mexico pharmacy
medication from mexico pharmacy: mexico pharmacy – medication from mexico pharmacy
https://mexicanpharmacy1st.com/# medicine in mexico pharmacies
best online pharmacies in mexico: mexican border pharmacies shipping to usa – mexican border pharmacies shipping to usa
mexico drug stores pharmacies mexico drug stores pharmacies mexican border pharmacies shipping to usa
buying from online mexican pharmacy: mexico pharmacies prescription drugs – mexican drugstore online
medicine in mexico pharmacies mexico pharmacy mexican border pharmacies shipping to usa
mexican border pharmacies shipping to usa: mexican mail order pharmacies – medication from mexico pharmacy
https://mexicanpharmacy1st.shop/# best online pharmacies in mexico
mexican rx online: buying prescription drugs in mexico online – best mexican online pharmacies
medicine in mexico pharmacies mexico pharmacies prescription drugs mexican mail order pharmacies
https://mexicanpharmacy1st.shop/# medication from mexico pharmacy
buying from online mexican pharmacy: pharmacies in mexico that ship to usa – mexican drugstore online
mexican pharmacy medication from mexico pharmacy mexico drug stores pharmacies
medicine in mexico pharmacies: mexican drugstore online – mexico pharmacies prescription drugs
https://mexicanpharmacy1st.online/# reputable mexican pharmacies online
mexican pharmaceuticals online: medication from mexico pharmacy – mexico pharmacies prescription drugs
mexico drug stores pharmacies medication from mexico pharmacy reputable mexican pharmacies online
reputable mexican pharmacies online mexico drug stores pharmacies mexican drugstore online
https://mexicanpharmacy1st.com/# п»їbest mexican online pharmacies
https://mexicanpharmacy1st.com/# mexico pharmacies prescription drugs
https://mexicanpharmacy1st.shop/# buying from online mexican pharmacy
buying prescription drugs in mexico: mexican pharmacy – purple pharmacy mexico price list
buying prescription drugs in mexico reputable mexican pharmacies online mexican border pharmacies shipping to usa
mexico pharmacy mexico pharmacy mexico pharmacy
https://mexicanpharmacy1st.online/# mexico drug stores pharmacies
buying prescription drugs in mexico online: mexican border pharmacies shipping to usa – mexican drugstore online
reputable mexican pharmacies online: mexican border pharmacies shipping to usa – medication from mexico pharmacy
https://mexicanpharmacy1st.online/# mexico drug stores pharmacies
mexico pharmacy: mexico pharmacy – best mexican online pharmacies
purple pharmacy mexico price list: mexican rx online – mexican rx online
purple pharmacy mexico price list mexico drug stores pharmacies buying from online mexican pharmacy
mexico drug stores pharmacies medicine in mexico pharmacies buying prescription drugs in mexico
https://lisinopril.club/# lisinopril generic over the counter
can i order generic clomid without rx: get generic clomid – can i purchase cheap clomid prices
order cheap propecia pill buying generic propecia without insurance propecia without rx
cost clomid price can i get cheap clomid tablets cost cheap clomid online
https://clomiphene.shop/# clomid without insurance
https://gabapentin.club/# gabapentin
lisinopril 10 mg tablet price: zestril 5mg – zestoretic price
https://gabapentin.club/# buying neurontin online
https://lisinopril.club/# zestril 2.5 mg
Cytotec 200mcg price: cytotec online – buy cytotec pills online cheap
lisinopril 25 mg lisinopril 3973 lisinopril 10mg tablet
can i buy clomid prices can you buy cheap clomid no prescription where to get generic clomid without rx
cytotec pills buy online: buy cytotec over the counter – cytotec online
get propecia price: buy cheap propecia no prescription – generic propecia without rx
neurontin 100mg capsule price neurontin cost generic neurontin generic brand
lisinopril 10 mg 12.5mg lisinopril 20 mg discount lisinopril 20 mg purchase
cost of generic propecia price: cost propecia – get propecia tablets
Fantastic read. I observed your internet site from a google search, and was glad i did. The data has helped me immensely.
http://cytotec.xyz/# cytotec pills online
http://gabapentin.club/# neurontin 600
how much is neurontin: neurontin tablets 300 mg – how to get neurontin cheap
https://gabapentin.club/# neurontin cost generic
best online pharmacy to buy ambien
cytotec abortion pill: buy cytotec over the counter – buy misoprostol over the counter
get generic propecia without insurance propecia cost cheap propecia price
https://lisinopril.club/# lisinopril 40 mg daily
get generic propecia tablets: cost of propecia without insurance – order generic propecia without rx
http://clomiphene.shop/# buying clomid without prescription
neurontin 800 mg tablets neurontin 202 neurontin 300 mg coupon
neurontin 100mg tablets neurontin medication neurontin 600 mg cost
cost clomid without insurance: get clomid without a prescription – cheap clomid pill
http://cytotec.xyz/# buy cytotec in usa
get generic propecia: cheap propecia without a prescription – buying propecia without insurance
https://lisinopril.club/# lisinopril mexico
price of lisinopril generic lisinopril 20 mg discount lisinopril cheap price
where to buy cheap clomid without dr prescription can i order generic clomid pill get generic clomid
http://clomiphene.shop/# can you get generic clomid tablets
can i purchase generic clomid pills: can you buy cheap clomid without prescription – cost of generic clomid tablets
http://gabapentin.club/# neurontin 600
neurontin 600 mg pill: cost of neurontin 100mg – buy generic neurontin online
http://propeciaf.online/# cost propecia without a prescription
cost of lisinopril 20 mg lisinopril 10 mg daily buy lisinopril 5 mg
buy misoprostol over the counter: buy misoprostol over the counter – cytotec abortion pill
cheap propecia for sale buy cheap propecia pill cheap propecia without prescription
buy cytotec in usa: cytotec buy online usa – buy cytotec over the counter
neurontin 100 mg cap: gabapentin buy – neurontin prices generic
buying propecia without a prescription: order propecia without prescription – cost of propecia without insurance
can i buy lisinopril online lisinopril 10 zestril 10 mg
cost of generic clomid without prescription how to get generic clomid online can i purchase clomid price
lisinopril online pharmacy: prices for lisinopril – lisinopril online purchase
https://clomiphene.shop/# can i buy generic clomid
zestoretic coupon: 30mg lisinopril – lisinopril 10mg prices compare
buy cytotec pills cytotec online cytotec pills buy online
buy cytotec pills online cheap buy misoprostol over the counter cytotec pills buy online
http://cheapestindia.com/# top online pharmacy india
best canadian online pharmacy: cheapestcanada.com – best canadian pharmacy to buy from
rxpharmacycoupons no prescription required pharmacy reputable online pharmacy no prescription
https://cheapestcanada.shop/# canadian pharmacy sarasota
http://cheapestmexico.com/# buying from online mexican pharmacy
https://36and6health.com/# pharmacy coupons
https://cheapestindia.com/# best india pharmacy
http://36and6health.com/# cheapest pharmacy to fill prescriptions without insurance
https://cheapestcanada.shop/# canadian neighbor pharmacy
https://cheapestcanada.com/# ed drugs online from canada
best canadian pharmacy cheapestcanada.com canada drugs reviews
http://cheapestindia.com/# top online pharmacy india
canadian pharmacy sarasota: cheapest canada – canadian pharmacy meds reviews
http://cheapestindia.com/# mail order pharmacy india
http://cheapestmexico.com/# best online pharmacies in mexico
https://cheapestcanada.com/# canadian pharmacy store
http://cheapestcanada.com/# online canadian pharmacy
https://cheapestindia.com/# india pharmacy mail order
https://cheapestindia.shop/# world pharmacy india
http://cheapestmexico.com/# mexican pharmaceuticals online
https://cheapestmexico.shop/# mexican mail order pharmacies
https://36and6health.shop/# online pharmacy no prescription needed
best online pharmacy india online pharmacy india Online medicine home delivery
Online medicine order: п»їlegitimate online pharmacies india – online pharmacy india
https://36and6health.com/# reputable online pharmacy no prescription
https://cheapestmexico.com/# mexican online pharmacies prescription drugs
http://cheapestindia.com/# best india pharmacy
https://36and6health.shop/# promo code for canadian pharmacy meds
https://eufarmacieonline.shop/# farmacie online affidabili
internet apotheke: shop apotheke gutschein – shop apotheke gutschein
Pharmacie sans ordonnance: pharmacie en ligne sans ordonnance – pharmacie en ligne sans ordonnance
farmacia online madrid farmacia online barata y fiable farmacia online espaГ±a envГo internacional
farmacia online barata y fiable: farmacia online espaГ±a envГo internacional – п»їfarmacia online espaГ±a
internet apotheke: online apotheke deutschland – eu apotheke ohne rezept
vente de mГ©dicament en ligne: Pharmacie en ligne livraison Europe – pharmacie en ligne
pharmacie en ligne france livraison belgique: pharmacie en ligne france fiable – Pharmacie Internationale en ligne
migliori farmacie online 2024: Farmacia online miglior prezzo – Farmacie online sicure
farmaci senza ricetta elenco farmacia online farmacia online
pharmacie en ligne france livraison belgique: pharmacie en ligne avec ordonnance – Pharmacie sans ordonnance
farmacie online sicure: Farmacia online migliore – Farmacia online più conveniente
farmacia online espaГ±a envГo internacional farmacia online envГo gratis farmacia online envГo gratis
farmacie online autorizzate elenco: migliori farmacie online 2024 – п»їFarmacia online migliore
acquisto farmaci con ricetta: п»їFarmacia online migliore – top farmacia online
farmacia online 24 horas: farmacia online envÃo gratis – farmacia online barata y fiable
online apotheke gГјnstig gГјnstigste online apotheke europa apotheke
farmacias online baratas: farmacias direct – farmacia online madrid
pharmacie en ligne avec ordonnance Pharmacie sans ordonnance п»їpharmacie en ligne france
https://eufarmacieonline.shop/# farmacia online piГ№ conveniente
п»їshop apotheke gutschein: gГјnstigste online apotheke – medikamente rezeptfrei
farmacia online españa envÃo internacional: farmacia online barata – farmacia online envÃo gratis
farmacie online autorizzate elenco: farmacie online sicure – farmacia online piГ№ conveniente
farmacia online barata farmacia online madrid farmacia online madrid
pharmacie en ligne france pas cher: pharmacie en ligne sans ordonnance – Achat mГ©dicament en ligne fiable
acheter médicament en ligne sans ordonnance: pharmacie en ligne france – trouver un médicament en pharmacie
pharmacie en ligne livraison europe Achat mГ©dicament en ligne fiable Pharmacie Internationale en ligne
acquisto farmaci con ricetta: migliori farmacie online 2024 – farmaci senza ricetta elenco
pharmacie en ligne france pas cher: pharmacie en ligne fiable – vente de mГ©dicament en ligne
farmacie online autorizzate elenco comprare farmaci online con ricetta п»їFarmacia online migliore
farmacia online barata y fiable: farmacias online seguras en espaГ±a – farmacias online seguras
internet apotheke: apotheke online – europa apotheke
apotheke online: online apotheke günstig – günstigste online apotheke
trouver un mГ©dicament en pharmacie pharmacie en ligne avec ordonnance acheter mГ©dicament en ligne sans ordonnance
eu apotheke ohne rezept: online apotheke rezept – gГјnstige online apotheke
vente de m̩dicament en ligne: Pharmacie sans ordonnance Рpharmacies en ligne certifi̩es
pharmacie en ligne fiable: vente de mГ©dicament en ligne – Pharmacie en ligne livraison Europe
apotheke online: ohne rezept apotheke – internet apotheke
http://eufarmaciaonline.com/# farmacia online envГo gratis
http://eumedicamentenligne.com/# Pharmacie en ligne livraison Europe
online apotheke deutschland: online apotheke günstig – online apotheke rezept
gГјnstigste online apotheke online apotheke deutschland gГјnstigste online apotheke
farmacia online barata: farmacias online seguras en espaГ±a – farmacia online barcelona
farmacia online barata: farmacia online envÃo gratis – farmacia online envÃo gratis
comprare farmaci online con ricetta: Farmacia online piГ№ conveniente – п»їFarmacia online migliore
farmacias online seguras en espaГ±a: farmacia en casa online descuento – farmacias online baratas
п»їFarmacia online migliore migliori farmacie online 2024 Farmacie on line spedizione gratuita
farmacia online madrid: farmacias online seguras – farmacia online barata
farmacias online baratas: farmacias online seguras – farmacia barata
comprare farmaci online con ricetta farmaci senza ricetta elenco acquisto farmaci con ricetta
acquisto farmaci con ricetta: Farmacie online sicure – Farmacia online migliore
beste online-apotheke ohne rezept: apotheke online – online apotheke rezept
gГјnstigste online apotheke medikament ohne rezept notfall gГјnstigste online apotheke
farmacias direct: farmacias online seguras – farmacia online madrid
Farmacia online miglior prezzo: top farmacia online – Farmacia online miglior prezzo
Amazing post – Gulvafslibning | Kurt Gulvmand however hey I having a issue we can not seem to be capable of sign up your rss feed, I am using bing reader fyi Thx ! Flash Factory
gГјnstigste online apotheke gГјnstige online apotheke gГјnstigste online apotheke
europa apotheke: medikament ohne rezept notfall – ohne rezept apotheke
https://eufarmacieonline.com/# farmaci senza ricetta elenco
http://eumedicamentenligne.com/# vente de mГ©dicament en ligne
Farmacie on line spedizione gratuita: farmacia online piГ№ conveniente – farmacie online sicure
farmacias online seguras en espaГ±a: farmacia en casa online descuento – farmacias online seguras en espaГ±a
online apotheke rezept online apotheke rezept medikamente rezeptfrei
farmacias online seguras: farmacia online madrid – farmacia online barata y fiable
acheter mГ©dicament en ligne sans ordonnance: acheter mГ©dicament en ligne sans ordonnance – pharmacie en ligne avec ordonnance
günstigste online apotheke: ohne rezept apotheke – internet apotheke
Pharmacie Internationale en ligne: Pharmacie Internationale en ligne – pharmacie en ligne sans ordonnance
Pharmacie en ligne livraison Europe pharmacie en ligne france fiable pharmacie en ligne fiable
top farmacia online: farmacie online affidabili – farmacia online
pharmacies en ligne certifiГ©es: trouver un mГ©dicament en pharmacie – pharmacie en ligne
online apotheke rezept: internet apotheke – online apotheke deutschland
farmacie online affidabili Farmacia online piГ№ conveniente farmacie online autorizzate elenco
migliori farmacie online 2024: comprare farmaci online all’estero – Farmacie on line spedizione gratuita
farmacie online autorizzate elenco migliori farmacie online 2024 farmacie online autorizzate elenco
internet apotheke: online apotheke gГјnstig – online apotheke deutschland
günstigste online apotheke: online apotheke rezept – beste online-apotheke ohne rezept
gГјnstige online apotheke: п»їshop apotheke gutschein – internet apotheke
Acheter Sildenafil 100mg sans ordonnance: Viagra sans ordonnance 24h suisse – Viagra femme sans ordonnance 24h
https://kamagraenligne.shop/# pharmacies en ligne certifiées
pharmacie en ligne Levitra acheter trouver un mГ©dicament en pharmacie
п»їpharmacie en ligne france levitra en ligne trouver un mГ©dicament en pharmacie
pharmacie en ligne avec ordonnance: kamagra 100mg prix – pharmacie en ligne france livraison internationale
trouver un mГ©dicament en pharmacie: kamagra livraison 24h – pharmacie en ligne france fiable
Pharmacie Internationale en ligne: levitra en ligne – Pharmacie en ligne livraison Europe
pharmacie en ligne france livraison internationale: Medicaments en ligne livres en 24h – Pharmacie sans ordonnance
Achat mГ©dicament en ligne fiable: levitra generique – vente de mГ©dicament en ligne
pharmacie en ligne pas cher: Cialis sans ordonnance 24h – acheter mГ©dicament en ligne sans ordonnance
pharmacies en ligne certifiГ©es: Medicaments en ligne livres en 24h – Pharmacie en ligne livraison Europe
acheter mГ©dicament en ligne sans ordonnance: Levitra pharmacie en ligne – pharmacie en ligne france pas cher
Viagra homme prix en pharmacie sans ordonnance: Quand une femme prend du Viagra homme – Viagra pas cher paris
Pharmacie Internationale en ligne: cialis generique – pharmacie en ligne fiable
https://cenligne.com/# pharmacie en ligne livraison europe
Viagra homme prix en pharmacie sans ordonnance: Viagra vente libre pays – Viagra sans ordonnance pharmacie France
vente de mГ©dicament en ligne: trouver un mГ©dicament en pharmacie – pharmacies en ligne certifiГ©es
Have you tried twitterfeed on your blog, i think it would be cool.”*.“
pharmacie en ligne france livraison belgique kamagra pas cher pharmacie en ligne fiable
Pharmacie en ligne livraison Europe: pharmacie en ligne france livraison belgique – pharmacie en ligne avec ordonnance
trouver un mГ©dicament en pharmacie: pharmacie en ligne france pas cher – pharmacie en ligne livraison europe
Pharmacie sans ordonnance: Cialis sans ordonnance 24h – pharmacie en ligne france fiable
pharmacie en ligne france fiable: levitra generique sites surs – Pharmacie Internationale en ligne
vente de mГ©dicament en ligne: Acheter Cialis 20 mg pas cher – vente de mГ©dicament en ligne
Hello.
This post was created with XRumer 23 StrongAI.
Good luck 🙂
http://cenligne.com/# pharmacie en ligne avec ordonnance
trouver un mГ©dicament en pharmacie: Pharmacie sans ordonnance – Pharmacie Internationale en ligne
Hello!
This post was created with XRumer 23 StrongAI.
Good luck 🙂
Pharmacie sans ordonnance: levitra generique prix en pharmacie – п»їpharmacie en ligne france
Pharmacie sans ordonnance: achat kamagra – Pharmacie en ligne livraison Europe
SildГ©nafil 100mg pharmacie en ligne: viagra en ligne – Viagra sans ordonnance livraison 48h
pharmacie en ligne sans ordonnance: cialis prix – trouver un mГ©dicament en pharmacie
pharmacie en ligne france livraison internationale: Levitra pharmacie en ligne – Pharmacie en ligne livraison Europe
pharmacie en ligne avec ordonnance: acheter kamagra site fiable – pharmacie en ligne
Pharmacie en ligne livraison Europe: Acheter Cialis 20 mg pas cher – pharmacie en ligne france fiable
Viagra gГ©nГ©rique pas cher livraison rapide: viagra sans ordonnance – Viagra homme prix en pharmacie sans ordonnance
I always used to read post in news papers but now as I am a user
of web thus from now I am using net for content, thanks to web.
pharmacie en ligne avec ordonnance: kamagra pas cher – pharmacie en ligne livraison europe
What a data of un-ambiguity and preserveness
of valuable know-how concerning unexpected feelings.
https://kamagraenligne.com/# Pharmacie en ligne livraison Europe
pharmacies en ligne certifiГ©es: Acheter Cialis 20 mg pas cher – pharmacie en ligne france livraison belgique
Viagra gГ©nГ©rique sans ordonnance en pharmacie: viagra en ligne – Viagra en france livraison rapide
Please let me know if you’re looking for a author for your weblog.
You have some really great posts and I believe I
would be a good asset. If you ever want to take some of the load off, I’d love
to write some articles for your blog in exchange for a link back to mine.
Please shoot me an e-mail if interested. Thanks!
This is my first time pay a quick visit
at here and i am truly pleassant to read everthing at single place.
Can I simply say what a comfort to discover someone that truly understands what
they’re discussing online. You actually understand how to bring an issue to
light and make it important. More people ought to check this out and understand
this side of your story. I was surprised that you aren’t more popular since you surely possess the gift.
vente de mГ©dicament en ligne: acheter kamagra site fiable – pharmacie en ligne livraison europe
Excellent goods from you, man. I’ve have in mind your stuff prior to and you are simply extremely fantastic.
I actually like what you’ve acquired right here, really like what you are
stating and the way during which you are saying it. You make it entertaining and you still care for
to keep it smart. I can’t wait to read far more from you.
This is actually a great web site.
Pharmacie sans ordonnance: kamagra livraison 24h – pharmacie en ligne sans ordonnance
Hmm is anyone else having problems with the images on this blog
loading? I’m trying to determine if its a problem on my end or if
it’s the blog. Any suggestions would be greatly appreciated.
Pharmacie en ligne livraison Europe: cialis sans ordonnance – trouver un mГ©dicament en pharmacie
pharmacie en ligne france livraison internationale: levitra generique sites surs – pharmacie en ligne france livraison internationale
Viagra femme ou trouver: Viagra sans ordonnance 24h – Viagra homme sans prescription
This paragraph will assist the internet visitors for building up new web site or even a weblog from start to end.
trouver un mГ©dicament en pharmacie: Levitra 20mg prix en pharmacie – pharmacie en ligne pas cher
pharmacie en ligne pas cher: kamagra en ligne – pharmacie en ligne sans ordonnance
Hi all, here every one is sharing these know-how,
therefore it’s nice to read this blog, and I used to pay a quick visit this website daily.
You made some really good points there. I looked on the net for
additional information about the issue and found most people will go along with your views on this
site.
I used to be suggested this website by my cousin. I’m no
longer certain whether or not this publish is written by him as nobody else know
such specific approximately my problem. You’re amazing!
Thank you!
Hurrah! Finally I got a webpage from where I know how to
truly obtain valuable facts concerning my study and knowledge.
vente de mГ©dicament en ligne: levitra generique sites surs – pharmacies en ligne certifiГ©es
acheter mГ©dicament en ligne sans ordonnance: Levitra pharmacie en ligne – acheter mГ©dicament en ligne sans ordonnance
pharmacie en ligne sans ordonnance: pharmacie en ligne – п»їpharmacie en ligne france
pharmacie en ligne sans ordonnance: pharmacie en ligne avec ordonnance – pharmacie en ligne france pas cher
acheter mГ©dicament en ligne sans ordonnance: Medicaments en ligne livres en 24h – Achat mГ©dicament en ligne fiable
Admiring the time and energy you put into
your website and detailed information you present.
It’s great to come across a blog every once in a while that isn’t
the same outdated rehashed material. Great read! I’ve bookmarked
your site and I’m adding your RSS feeds to
my Google account.
Pharmacie sans ordonnance: cialis sans ordonnance – Pharmacie sans ordonnance
acheter mГ©dicament en ligne sans ordonnance: Pharmacies en ligne certifiees – Pharmacie en ligne livraison Europe
It’s actually very complicated in this busy life to listen news
on TV, therefore I just use internet for that reason, and get the most recent news.
Ahaa, its pleasant dialogue about this piece of writing at this
place at this blog, I have read all that, so now me
also commenting here.
Good day! This is kind of off topic but I need some help from
an established blog. Is it tough to set up your
own blog? I’m not very techincal but I can figure things out pretty fast.
I’m thinking about setting up my own but I’m not sure where to begin. Do you have any points or suggestions?
Many thanks
Viagra homme prix en pharmacie sans ordonnance: viagra sans ordonnance – Viagra homme prix en pharmacie sans ordonnance
Viagra pas cher inde: viagra en ligne – Viagra gГ©nГ©rique pas cher livraison rapide
Good site you’ve got here.. It’s difficult to find high-quality
writing like yours these days. I truly appreciate
people like you! Take care!!
I loved as much as you’ll receive carried out right here.
The sketch is tasteful, your authored subject matter stylish.
nonetheless, you command get got an impatience over that you
wish be delivering the following. unwell unquestionably come further formerly again since exactly the same nearly very often inside
case you shield this hike.
pharmacie en ligne: kamagra oral jelly – pharmacie en ligne france pas cher
Does your site have a contact page? I’m having
a tough time locating it but, I’d like to shoot you an e-mail.
I’ve got some creative ideas for your blog you might be
interested in hearing. Either way, great blog and I look forward to seeing it develop over time.
Viagra sans ordonnance livraison 48h: viagra en ligne – Viagra femme sans ordonnance 24h
Achat mГ©dicament en ligne fiable: cialis sans ordonnance – acheter mГ©dicament en ligne sans ordonnance
pharmacie en ligne livraison europe: cialis generique – pharmacie en ligne france fiable
I used to be able to find good advice from your articles.
Hi to every , since I am in fact keen of reading this weblog’s post to be
updated on a regular basis. It contains fastidious stuff.
pharmacie en ligne sans ordonnance: pharmacie en ligne avec ordonnance – vente de mГ©dicament en ligne
Viagra pas cher livraison rapide france: Viagra generique en pharmacie – Viagra 100 mg sans ordonnance
Pharmacie en ligne livraison Europe: Levitra 20mg prix en pharmacie – acheter mГ©dicament en ligne sans ordonnance
Attractive section of content. I just stumbled upon your web site and in accession capital to assert that I acquire
actually enjoyed account your blog posts. Any way I will be subscribing to your augment and even I achievement you access consistently quickly.
Quand une femme prend du Viagra homme: Viagra sans ordonnance 24h – SildГ©nafil Teva 100 mg acheter
pharmacie en ligne livraison europe: Pharmacies en ligne certifiees – Pharmacie Internationale en ligne
pharmacie en ligne france livraison belgique: levitra en ligne – pharmacie en ligne france fiable
pharmacie en ligne france livraison internationale: levitra generique sites surs – pharmacies en ligne certifiГ©es
pharmacie en ligne france pas cher: Levitra pharmacie en ligne – pharmacie en ligne pas cher
vente de mГ©dicament en ligne: cialis generique – Pharmacie en ligne livraison Europe
pharmacie en ligne: Pharmacies en ligne certifiees – pharmacies en ligne certifiГ©es
Viagra sans ordonnance 24h suisse: Acheter du Viagra sans ordonnance – Viagra sans ordonnance pharmacie France
trouver un mГ©dicament en pharmacie: cialis prix – vente de mГ©dicament en ligne
trouver un mГ©dicament en pharmacie: pharmacie en ligne sans ordonnance – pharmacie en ligne france livraison internationale
pharmacie en ligne france livraison internationale: cialis generique – Achat mГ©dicament en ligne fiable
pharmacie en ligne pas cher: kamagra oral jelly – trouver un mГ©dicament en pharmacie
pharmacie en ligne france livraison internationale: pharmacie en ligne france livraison internationale – Achat mГ©dicament en ligne fiable
Achat mГ©dicament en ligne fiable: Cialis sans ordonnance 24h – pharmacie en ligne livraison europe
trouver un mГ©dicament en pharmacie: levitra generique prix en pharmacie – pharmacie en ligne france pas cher
vente de mГ©dicament en ligne: kamagra gel – acheter mГ©dicament en ligne sans ordonnance
Viagra pas cher livraison rapide france: Viagra generique en pharmacie – Viagra 100 mg sans ordonnance
Achat mГ©dicament en ligne fiable: acheter kamagra site fiable – Pharmacie sans ordonnance
Quand une femme prend du Viagra homme: Meilleur Viagra sans ordonnance 24h – Viagra pas cher paris
pharmacie en ligne france livraison internationale: Acheter Cialis – pharmacies en ligne certifiГ©es
pharmacie en ligne france livraison belgique: levitra generique sites surs – pharmacie en ligne livraison europe
Viagra homme prix en pharmacie sans ordonnance: Viagra generique en pharmacie – Viagra sans ordonnance livraison 48h
http://kamagraenligne.com/# Achat médicament en ligne fiable
trouver un mГ©dicament en pharmacie: levitra generique sites surs – acheter mГ©dicament en ligne sans ordonnance
Pharmacie Internationale en ligne: cialis prix – pharmacies en ligne certifiГ©es
Pharmacie Internationale en ligne: Acheter Cialis 20 mg pas cher – Pharmacie en ligne livraison Europe
Achat mГ©dicament en ligne fiable: kamagra en ligne – pharmacie en ligne france fiable
pharmacies en ligne certifiГ©es: achat kamagra – Achat mГ©dicament en ligne fiable
Achat mГ©dicament en ligne fiable: kamagra livraison 24h – pharmacie en ligne livraison europe
pharmacie en ligne sans ordonnance: kamagra en ligne – pharmacie en ligne france livraison belgique
pharmacies en ligne certifiГ©es: Levitra 20mg prix en pharmacie – Achat mГ©dicament en ligne fiable
SildГ©nafil 100 mg prix en pharmacie en France: viagra sans ordonnance – Acheter viagra en ligne livraison 24h
trouver un mГ©dicament en pharmacie: kamagra 100mg prix – pharmacie en ligne sans ordonnance
pharmacie en ligne fiable: kamagra livraison 24h – trouver un mГ©dicament en pharmacie
pharmacie en ligne avec ordonnance: pharmacie en ligne sans ordonnance – п»їpharmacie en ligne france
Pharmacie Internationale en ligne: pharmacie en ligne pas cher – pharmacie en ligne france pas cher
acheter mГ©dicament en ligne sans ordonnance: pharmacie en ligne pas cher – pharmacies en ligne certifiГ©es
pharmacie en ligne avec ordonnance: pharmacies en ligne certifiГ©es – pharmacie en ligne avec ordonnance
pharmacie en ligne france livraison belgique: Cialis sans ordonnance 24h – pharmacie en ligne sans ordonnance
acheter mГ©dicament en ligne sans ordonnance: Pharmacies en ligne certifiees – Pharmacie en ligne livraison Europe
vente de mГ©dicament en ligne: Medicaments en ligne livres en 24h – vente de mГ©dicament en ligne
When someone writes an piece of writing he/she keeps the plan of a user in his/her mind that how
a user can understand it. Thus that’s why this paragraph
is outstdanding. Thanks!
geinoutime.com
그래서 그는 급히 말했습니다. “아버지, 저들이…”
He never realized how far-reaching this one decision would be. I read texts that they wrote to each other while we were still married saying
frog the gambler is the essential resource for value-conscious sports-bettors with an interest in optimising their betting performance and beating the sportsbooks. KickOff is the home of free football betting tips. Here you’ll find free tips for every major European football league and competition. We give you simple to use, detailed data, and tips on a daily basis, to help with your betting strategy. You’ll find today’s fixtures as well as mid-week and weekend fixtures. We suppose to see many goals and so at least 3 strikes should not be high limit. We really appreciate this good odd for this high winning probability. You might be wondering how we make our football predictions. Crucially, we combine data and knowledge of all teams and leagues to create the best football tips tomorrow can boast. Also, we take our time to analyse the data we have to give our players the best possible predictions. Our prediction results are based on both statistics and research.
https://keluaranharian.xyz/mengetahui-macam-permainan-slot-everlasting-spins/
Are you an enthusiastic sports betting fan that is is always looking the best odds like the early bird that catches the worm? Worry not. We’ve got you covered! If you are simply looking for the best prices for tomorrow’s games, we will help you out. In this section, you will get all the stats, odds, analysis and betting tips that will fulfil your needs.SPECIAL ADVISE: Take a closer look at our betting bonus section to find a fitting welcome offer. Wherever you are, SportsGrid can be there too. Here are all the platforms and subscription services you can find us on: Wondering which football team to bet on tomorrow? Freesupertips has got you covered. Our experts work days ahead of the sporting calendar to make sure they are able to publish their football predictions well ahead of the day of the match. We cover all the major european leagues and various other popular football leagues around the world.
geinoutime.com
일어선 것은 온통 얼굴에 뾰루지가 난 거친 남자였다.
What is the role of the FDA in ensuring the safety of medicines used in the treatment of rare genetic disorders cenforce 200 werking?
п»їpharmacie en ligne france: pharmacie en ligne pas cher – pharmacie en ligne livraison europe
pharmacie en ligne pas cher https://kamagraenligne.com/# pharmacie en ligne avec ordonnance
cheap amoxicillin 500mg: amoxil best price – cheap amoxicillin 500mg
https://amoxicillinca.com/# zithromax 500 mg lowest price drugstore online
where to buy generic clomid now clomid Prednisonerxa how to buy clomid no prescription
cost doxycycline: doxycycline best price – buy 40 mg doxycycline
zithromax for sale us: cheapest Azithromycin – how to buy zithromax online
https://clomidca.com/# generic prednisone pills
zithromax antibiotic: Azithromycin – buy zithromax without prescription online
http://doxycyclineca.com/# amoxicillin generic
how can i get doxycycline over the counter: doxycycline – can i buy doxycycline over the counter in europe
fast shipping prednisone buy online prednisone 20mg by mail order
cheap amoxicillin 500mg: amoxil online – can i buy amoxicillin online
https://amoxicillinca.com/# buy zithromax online australia
doxycycline gel: buy tetracycline antibiotics – doxycycline 40 mg cost
amoxicillin without rx: cheapest amoxicillin – amoxicillin 500 mg without a prescription
how to order doxycycline azithromycinca doxycycline generic price
https://azithromycinca.shop/# doxycycline prescription online
buy amoxicillin 500mg usa: amoxil – amoxicillin without prescription
https://azithromycinca.com/# doxycycline 75 mg
prednisone 2.5 mg daily: Steroid – prednisone oral
can i order cheap clomid tablets Prednisonerxa clomid medication
can i buy generic clomid pills: cheap fertility drug – cheap clomid
https://clomidca.com/# iv prednisone
buying prednisone mexico: prednisone – otc prednisone cream
http://clomidca.com/# prednisone prescription online
buy cheap generic zithromax: cheapest Azithromycin – zithromax 500 price
amoxicillin 500 amoxil rexall pharmacy amoxicillin 500mg
zithromax 500 mg lowest price drugstore online: Azithromycin – can you buy zithromax over the counter in australia
http://prednisonerxa.com/# can i order generic clomid
how can i get prednisone: prednisone clomidca – prednisone daily
purchase zithromax z-pak: buy zithromax online – zithromax pill
https://clomidca.shop/# prednisone 10 mg price
doxycycline purchase azithromycinca.com doxycycline brand name
where to get prednisone: Deltasone – prednisone 50
https://prednisonerxa.com/# clomid rx
can i get generic clomid without dr prescription: best price – cheap clomid price
prednisone cost in india: clomidca.com – prednisone
50 mg prednisone from canada Deltasone prednisone 10mg buy online
http://clomidca.com/# prednisone 5084
doxycycline 50mg tab: doxycycline best price – how much is doxycycline 100mg
where to get clomid now: clomid – where to buy generic clomid tablets
prednisone 20mg prescription cost: buy online – prednisone 60 mg tablet
https://prednisonerxa.com/# can i purchase cheap clomid no prescription
amoxicillin 500mg amoxil best price can i purchase amoxicillin online
zithromax over the counter uk: Azithromycin – zithromax 500
http://amoxicillinca.com/# buy zithromax 1000 mg online
amoxicillin 500mg prescription: amoxil online – amoxicillin pills 500 mg
how to buy doxycycline online: azithromycinca.shop – doxycycline capsules price in india
buy generic zithromax no prescription Azithromycin zithromax capsules price
https://clomidca.shop/# prednisone online india
amoxicillin 500mg price: amoxil – amoxicillin 500 mg brand name
http://clomidca.com/# prednisone 20 mg tablet
zithromax capsules price: buy zithromax online – zithromax order online uk
prednisone 10mg price in india: prednisone – how to get prednisone tablets
can you buy prednisone in canada clomidca prednisone 60 mg daily
http://azithromycinca.com/# doxycycline prescription uk
zithromax canadian pharmacy: zithromax – zithromax 250 mg tablet price
purchase zithromax z-pak: Azithromycin best price – zithromax for sale cheap
http://prednisonerxa.com/# where buy clomid for sale
amoxicillin 500 coupon amoxil doxycyclineca amoxicillin buy no prescription
can i order generic clomid without a prescription: prednisonerxa.shop – how to get generic clomid tablets
https://prednisonerxa.com/# cost cheap clomid no prescription
how to buy clomid without prescription: best price – cheap clomid pills
medicine doxycycline 100mg: azithromycinca.com – doxycycline online canada
https://amoxicillinca.shop/# zithromax capsules
buy generic zithromax no prescription buy zithromax online generic zithromax medicine
generic amoxicillin 500mg: amoxil online – over the counter amoxicillin
https://doxycyclineca.com/# azithromycin amoxicillin
zithromax tablets for sale: amoxicillinca – buy cheap zithromax online
generic zithromax online paypal: buy zithromax amoxicillinca – zithromax online pharmacy canada
buy doxycycline online usa doxycycline best price doxycycline india pharmacy
https://clomidca.com/# prednisone for sale in canada
doxycycline best price: doxycycline – where can i get doxycycline over the counter
1250 mg prednisone: clomidca.shop – prednisone daily use
https://doxycyclineca.shop/# buy amoxicillin online with paypal
buy prednisone tablets online: buy online – where to buy prednisone without prescription
cheap zithromax pills buy zithromax amoxicillinca can you buy zithromax over the counter
https://doxycyclineca.shop/# generic amoxicillin online
buy prednisone canada: buy online – prednisone 30 mg coupon
where to buy amoxicillin over the counter: doxycyclineca – how to get amoxicillin over the counter
http://prednisonerxa.com/# cost of generic clomid tablets
doxycycline singapore: doxycycline best price – doxycycline 631311
cheap amoxicillin 500mg amoxil online amoxicillin 500 mg capsule
https://prednisonerxa.shop/# how can i get cheap clomid without a prescription
zithromax antibiotic without prescription: Azithromycin best price – zithromax coupon
buy cheap clomid without prescription: clomid – get cheap clomid without dr prescription
https://clomidca.com/# purchase prednisone
zithromax 250mg cheapest Azithromycin zithromax 500 price
prednisone generic brand name: clomidca.com – steroids prednisone for sale
https://clomidca.com/# can i buy prednisone online in uk
amoxicillin in india: amoxil – amoxicillin generic brand
online order prednisone: clomidca – order prednisone on line
https://amoxicillinca.shop/# zithromax over the counter uk
buy doxycycline mexico azithromycinca.com doxycycline hyclate 100 mg
can i buy cheap clomid without prescription: Prednisonerxa – can you buy cheap clomid without rx
https://clomidca.shop/# prednisone purchase canada
how to buy zithromax online: cheapest Azithromycin – zithromax 500 tablet
amoxicillin 875 125 mg tab: amoxil online – cost of amoxicillin 875 mg
zithromax 250 mg zithromax can i buy zithromax online
https://prednisonerxa.com/# can i get cheap clomid no prescription
prednisone 10 mg price: Steroid – prednisone tablets 2.5 mg
buy zithromax online australia: Azithromycin – buy zithromax without presc
http://prednisonerxa.com/# where can i get generic clomid without insurance
amoxicillin 250 mg price in india: amoxil best price – buy amoxicillin canada
https://azithromycinca.shop/# doxycycline uk
zithromax 250 price buy zithromax online zithromax cost uk
cost clomid no prescription: clomid Prednisonerxa – how to get generic clomid pills
where to get clomid price: prednisonerxa.com – where can i get cheap clomid no prescription
https://prednisonerxa.shop/# clomid pill
order generic clomid prices: prednisonerxa.com – how can i get cheap clomid without insurance
http://clomidca.com/# fast shipping prednisone
generic amoxicillin over the counter amoxil online amoxicillin pills 500 mg
how to buy clomid for sale: prednisonerxa.shop – cost of cheap clomid without a prescription
doxycycline 100 mg cap over the counter: here – where can i buy doxycycline no prescription
https://azithromycinca.shop/# doxycycline mono
zithromax 500 mg lowest price pharmacy online: amoxicillinca – zithromax drug
https://clomidca.com/# prednisone 5093
where can i buy generic clomid now best price get generic clomid pills
zithromax canadian pharmacy: cheapest Azithromycin – where can i get zithromax
can i buy cheap clomid without prescription: clomid – how to get generic clomid without prescription
https://doxycyclineca.shop/# buy amoxicillin online uk
amoxicillin over counter: amoxil best price – amoxicillin 500 mg cost
amoxicillin brand name amoxil best price amoxicillin medicine over the counter
http://doxycyclineca.com/# can i purchase amoxicillin online
generic zithromax azithromycin: amoxicillinca – zithromax online usa no prescription
doxycycline cap price: doxycycline – doxycycline over the counter
https://amoxicillinca.shop/# zithromax prescription
amoxicillin 250 mg cheapest amoxicillin buy amoxicillin online mexico
azithromycin zithromax: zithromax – buy zithromax
https://amoxicillinca.com/# buy zithromax online fast shipping
discount doxycycline: doxycycline – where can i buy doxycycline
prescription for amoxicillin: amoxicillin – how to buy amoxycillin
https://amoxicillinca.com/# zithromax 600 mg tablets
can you get cheap clomid without insurance prednisonerxa.shop cheap clomid without dr prescription
doxycycline compare prices: doxycycline – doxycycline 500mg price
buy amoxicillin online without prescription: buy cheapest antibiotics – amoxicillin 875 mg tablet
https://azithromycinca.com/# cost of doxycycline in canada
doxycycline 100mg buy online: buy tetracycline antibiotics – doxycycline 100 mg
http://doxycyclineca.com/# buy amoxicillin online without prescription
buy amoxicillin buy cheapest antibiotics amoxicillin 500 mg tablet price
30mg prednisone: clomidca.shop – prednisone cost 10mg
amoxicillin cost australia: doxycyclineca – generic amoxicillin over the counter
https://clomidca.shop/# mail order prednisone
compare prednisone prices: Deltasone – prednisone 50
http://azithromycinca.com/# order doxycycline online australia
prednisone prescription drug clomidca.com buy prednisone without a prescription
30mg prednisone: buy online – prednisone online paypal
prednisone price australia: clomidca.shop – order prednisone 100g online without prescription
http://azithromycinca.com/# doxycycline 100mg capsules price
where to buy clomid no prescription: prednisonerxa.com – where to get clomid without insurance
http://amoxicillinca.com/# zithromax 500mg
generic zithromax azithromycin Azithromycin can you buy zithromax over the counter
can i buy zithromax over the counter in canada: buy zithromax online – where can i buy zithromax in canada
where to get generic clomid pills: best price – buying clomid pills
https://prednisonerxa.com/# can you get clomid
24 hour drug store
prednisolone prednisone: clomidca – prednisone online pharmacy
compare prednisone prices: clomidca.shop – brand prednisone
http://prednisonerxa.com/# how can i get cheap clomid no prescription
zithromax for sale us: buy zithromax amoxicillinca – zithromax over the counter canada
https://azithromycinca.com/# doxycycline 100mg tablet brand name
zithromax prescription online: Azithromycin best price – where can you buy zithromax
generic zithromax over the counter: buy zithromax online – zithromax cost uk
https://amoxicillinca.shop/# zithromax 250
zithromax 500mg price in india: cheapest Azithromycin – where can i get zithromax
https://prednisonerxa.shop/# clomid cheap
amoxicillin 500mg over the counter: buy cheapest antibiotics – amoxicillin order online no prescription
order prednisone with mastercard debit: clomidca.com – prednisone 40 mg price
doxycycline brand: here – doxycycline online sale
prednisone 15 mg daily: Deltasone – prednisone medicine
http://doxycyclineca.com/# medicine amoxicillin 500mg
canadian pharmacy amoxicillin: cheapest amoxicillin – amoxicillin buy no prescription
https://prednisonerxa.com/# generic clomid for sale
amoxicillin 500mg capsule cost: buy cheapest antibiotics – how much is amoxicillin
amoxicillin pharmacy price: amoxil best price – amoxicillin discount coupon
https://clomidca.shop/# prednisone 20 mg pill
zithromax order online uk: buy zithromax online – zithromax 1000 mg pills
https://amoxicillinca.com/# can you buy zithromax over the counter
doxycycline tablets over the counter: azithromycinca.com – doxycycline hyclate 100mg price
where to get cheap clomid price: prednisonerxa.com – can you buy clomid without a prescription
https://amoxicillinca.shop/# zithromax for sale 500 mg
amar pharmacy rx
doxycycline 5553: azithromycinca – where can i buy doxycycline
canada buy prednisone online: clomidca.shop – prednisone 20mg price in india
http://doxycyclineca.com/# amoxicillin generic
can i order generic clomid pills: best price – buying clomid prices
https://prednisonerxa.com/# buying generic clomid price
prednisone 60 mg tablet: clomidca.shop – prednisone uk over the counter
doxycycline mexico: doxycycline – doxycycline online with no prescription
https://clomidca.shop/# prednisone 5 mg tablet price
buy amoxicillin online uk: amoxil doxycyclineca – where to buy amoxicillin 500mg without prescription
https://amoxicillinca.com/# zithromax online usa
medicine amoxicillin 500: doxycyclineca – buy amoxicillin without prescription
amoxicillin online purchase
https://azithromycinca.shop/# doxylin
doxycycline 100mg capsule sale
pin-up360: Pin Up Azerbaycan – Pin up 306 casino
Enjoyed looking through this, very good stuff, appreciate it.
https://autolux-azerbaijan.com/# pin-up kazino
https://autolux-azerbaijan.com/# Pin Up Kazino ?Onlayn
Pin-Up Casino: ?Onlayn Kazino – pin-up kazino
Pin Up Azerbaycan: Pin-Up Casino – Pin-Up Casino
I am glad that I found this weblog , exactly the right info that I was searching for! .
https://autolux-azerbaijan.com/# Pin up 306 casino
Pin Up Kazino ?Onlayn: ?Onlayn Kazino – pin-up kazino
https://autolux-azerbaijan.com/# pin-up 141 casino
Pin up 306 casino: Pin-Up Casino – Pin Up Azerbaycan ?Onlayn Kazino
https://autolux-azerbaijan.com/# pin-up kazino
https://autolux-azerbaijan.com/# Pin Up Kazino ?Onlayn
pin-up kazino: pin-up kazino – Pin-Up Casino
https://autolux-azerbaijan.com/# pin-up360
https://autolux-azerbaijan.com/# Pin Up Azerbaycan
Pin-up Giris: Pin up 306 casino – pin-up kazino
Pin up 306 casino: Pin Up Kazino ?Onlayn – Pin Up Azerbaycan
https://autolux-azerbaijan.com/# pin-up360
https://northern-doctors.org/# mexican rx online
mexico drug stores pharmacies: buying prescription drugs in mexico – medication from mexico pharmacy
https://northern-doctors.org/# mexico pharmacies prescription drugs
п»їbest mexican online pharmacies mexican pharmacy buying prescription drugs in mexico
mexican rx online: northern doctors – mexican mail order pharmacies
http://northern-doctors.org/# medication from mexico pharmacy
https://northern-doctors.org/# п»їbest mexican online pharmacies
buying from online mexican pharmacy: mexican pharmacy northern doctors – buying prescription drugs in mexico online
mexico drug stores pharmacies Mexico pharmacy that ship to usa mexico drug stores pharmacies
mexican mail order pharmacies: mexican pharmacy – pharmacies in mexico that ship to usa
pharmacies in mexico that ship to usa: mexican pharmacy online – mexican pharmaceuticals online
https://northern-doctors.org/# reputable mexican pharmacies online
mexican online pharmacies prescription drugs: northern doctors – medicine in mexico pharmacies
best online pharmacies in mexico: mexican pharmacy – mexican drugstore online
mexican online pharmacies prescription drugs mexican pharmacy online medication from mexico pharmacy
mexico pharmacies prescription drugs: mexican pharmacy – mexico drug stores pharmacies
https://northern-doctors.org/# buying from online mexican pharmacy
mexico drug stores pharmacies: Mexico pharmacy that ship to usa – mexico pharmacies prescription drugs
reputable mexican pharmacies online: mexican northern doctors – pharmacies in mexico that ship to usa
https://northern-doctors.org/# mexico pharmacies prescription drugs
mexican online pharmacies prescription drugs: mexican pharmacy – mexican drugstore online
온라인 슬롯
그냥 향초였는데 품절이네요.2월 15일 그는 궁원에 정착했다.
mexico pharmacies prescription drugs: Mexico pharmacy that ship to usa – mexican drugstore online
https://northern-doctors.org/# mexican online pharmacies prescription drugs
mexican online pharmacies prescription drugs mexican northern doctors mexican drugstore online
mexico drug stores pharmacies: mexican pharmacy online – buying prescription drugs in mexico online
https://northern-doctors.org/# purple pharmacy mexico price list
mexican pharmaceuticals online: mexican pharmacy online – medicine in mexico pharmacies
buying from online mexican pharmacy Mexico pharmacy that ship to usa medicine in mexico pharmacies
http://northern-doctors.org/# buying prescription drugs in mexico
buying from online mexican pharmacy: northern doctors – mexican rx online
http://northern-doctors.org/# pharmacies in mexico that ship to usa
best online pharmacies in mexico: mexican online pharmacies prescription drugs – mexico pharmacy
https://northern-doctors.org/# medicine in mexico pharmacies
mexican border pharmacies shipping to usa: Mexico pharmacy that ship to usa – mexican border pharmacies shipping to usa
mexican pharmaceuticals online Mexico pharmacy that ship to usa buying prescription drugs in mexico online
mexican pharmaceuticals online: mexican pharmacy – reputable mexican pharmacies online
п»їbest mexican online pharmacies: northern doctors pharmacy – mexico drug stores pharmacies
https://northern-doctors.org/# buying prescription drugs in mexico online
mexico pharmacies prescription drugs: mexican pharmacy online – medicine in mexico pharmacies
http://northern-doctors.org/# best online pharmacies in mexico
buying prescription drugs in mexico: mexican pharmacy online – mexico drug stores pharmacies
п»їbest mexican online pharmacies: northern doctors pharmacy – best online pharmacies in mexico
mexican pharmacy: mexican pharmacy online – mexican pharmaceuticals online
mexican border pharmacies shipping to usa buying from online mexican pharmacy mexican online pharmacies prescription drugs
best online pharmacies in mexico: northern doctors – mexican rx online
mexican online pharmacies prescription drugs: northern doctors pharmacy – п»їbest mexican online pharmacies
https://northern-doctors.org/# buying from online mexican pharmacy
reputable mexican pharmacies online: mexican pharmacy online – mexico pharmacies prescription drugs
reputable mexican pharmacies online: mexican border pharmacies shipping to usa – pharmacies in mexico that ship to usa
https://northern-doctors.org/# pharmacies in mexico that ship to usa
http://northern-doctors.org/# mexican online pharmacies prescription drugs
mexican pharmacy: mexican pharmacy online – mexico pharmacies prescription drugs
buying prescription drugs in mexico online: mexican northern doctors – mexican online pharmacies prescription drugs
mexico pharmacy online mexican pharmacy mexico pharmacy
https://cmqpharma.com/# buying prescription drugs in mexico online
mexican pharmaceuticals online
buying from online mexican pharmacy mexican pharmacy mexican drugstore online
mexican border pharmacies shipping to usa mexico pharmacy reputable mexican pharmacies online
mexican border pharmacies shipping to usa cmq mexican pharmacy online pharmacies in mexico that ship to usa
mexican online pharmacies prescription drugs cmq mexican pharmacy online mexico pharmacy
mexican online pharmacies prescription drugs cmq mexican pharmacy online pharmacies in mexico that ship to usa
buying from online mexican pharmacy mexican online pharmacy mexican online pharmacies prescription drugs
mexico pharmacies prescription drugs cmq pharma buying from online mexican pharmacy
п»їbest mexican online pharmacies mexican pharmacy mexican border pharmacies shipping to usa
mexican border pharmacies shipping to usa cmqpharma.com mexican drugstore online
mexican drugstore online mexican pharmacy reputable mexican pharmacies online
http://cmqpharma.com/# medication from mexico pharmacy
reputable mexican pharmacies online
When I originally commented I clicked the -Notify me when new surveys are added- checkbox and today every time a comment is added I purchase four emails with similar comment. Perhaps there is however you possibly can get rid of me from that service? Thanks!
very nice post, i actually love this website, carry on it
buying from online mexican pharmacy: cmqpharma.com – mexico pharmacies prescription drugs
This site definitely has all of the information I needed about this subject
My website: russkoeporno365.pro
In the present competitive online casino industry , Turnkey Solution Software has emerged as a game-changer for operators looking to establish and grow their presence in the market. These comprehensive solutions offer everything from customizable platforms and extensive games portfolio to payment processing and customer support, providing operators with all the tools they need to launch and manage a successful online casino business. Our turnkey casino platform supports fiat as well as cryptocurrency payment options for ensuring smooth transactions. You can email the site owner to let them know you were blocked. Please include what you were doing when this page came up and the Cloudflare Ray ID found at the bottom of this page. © 1Click Games As we transition to our proprietary platform, we’re offering an exceptional opportunity to acquire our established White Label Casino Business. This package includes two highly successful casino… More details »
https://wiki-net.win/index.php?title=100_casino_welcome_bonus
While working your way through my latest online casino and betting site reviews, you’ll notice that I make frequent reference to “finding the best”. From an objective standpoint, this is completely true – I am always on the lookout for an online casino that provides the most complete experience. However, beyond the obvious desire to find exceptional legitimate and licenced online casinos, my role is to find you an experience that almost feels tailor-made. In this article, you’ll come across various PA online casino welcome packages that offer all sorts of bonuses to first-time players. You’ll also get a rundown of the types of promotions offered, some pros and cons of all of our top PA online casinos, and so much more. If you’re in search of information regarding any of the legal online casinos in Pennsylvania, you’ve come to the right place.
Thanks for sharing. I read many of your blog posts, cool, your blog is very good.
Major League Soccer side LA Galaxy are plotting a double swoop for former Borussia Dortmund pair Marco Reus and Robert Lewandowski. But how would I assign wins draws or losses? And calculate the goals scored and goals against? Ispe FC performance and form graph is a Sofascore unique algorithm that we are generating from the team’s last 10 matches, statistics, detailed analysis and our own knowledge. Teams 1. The Final Score Soccer Universe file did not include the top Guatemalan league (The 2nd Division is included, probably by a mistake). Thus I don’t have ratings for Comunicaciones or Municipal. To avoid this problem, I have moved them back to match their real-life group opponents and will use real life results for those matches, just like I said I would be doing for Alpha United of Guyana.
http://sports4.iceserver.co.kr/bbs/board.php?bo_table=free&wr_id=1203244
Liverpool lifted their fifth Champions League title at the stadium in 2005, after staging their now historic comeback when they found themselves 3-0 down at halftime to AC Milan. All times U.S. Eastern The players are in the tunnel. Manchester United are wearing their John Squire Jackson Pollock trackie tops. The combined band of the armed forces plays and there’s the worker bee, an emblem of the city of Manchester, on the podium. WinW FA Cup final 2024 LIVE: Manchester United subsitutes From De Bruyne’s incredible pass to Guimaraes’ outside-of-the-boot flick. Watch some of the most stunning assists of the campaign Wednesday, March 6 Real Madrid 1-1 (2-1 agg.) RB Leipzig – Recap, highlightsManchester City 3-1 (6-2 agg.) Copenhagen Manchester City won an unprecedented fourth straight English top flight title with a 3-1 win over West Ham United on Sunday to pip rivals Arsenal on the final day of a thrilling Premier League season as the fans turned the pitch into a sea of blue.
canadian pharmacy mall: pharmacy in canada – cross border pharmacy canada
pharmacy canadian canadian pharmacy meds review adderall canadian pharmacy
http://indiapharmast.com/# mail order pharmacy india
mexico drug stores pharmacies: mexico pharmacy – mexico drug stores pharmacies
mexico pharmacy: mexican drugstore online – buying prescription drugs in mexico online
canadianpharmacyworld com canadian pharmacy meds reviews cross border pharmacy canada
top 10 pharmacies in india: best india pharmacy – buy medicines online in india
northern pharmacy canada: safe canadian pharmacies – canadian pharmacy ratings
http://foruspharma.com/# mexico drug stores pharmacies
best online canadian pharmacy: reddit canadian pharmacy – northwest pharmacy canada
online pharmacy canada: www canadianonlinepharmacy – my canadian pharmacy
india pharmacy mail order mail order pharmacy india reputable indian online pharmacy
mail order pharmacy india: online shopping pharmacy india – pharmacy website india
http://indiapharmast.com/# world pharmacy india
top online pharmacy india: online pharmacy india – indian pharmacy online
indian pharmacy indian pharmacy world pharmacy india
canadian pharmacy com: legitimate canadian mail order pharmacy – canadian pharmacy service
canadian pharmacy near me: legal to buy prescription drugs from canada – canadian pharmacy online
How does the dosage change if I’m switching from a brand to a generic version avanafil 50 mg?
generic amoxicillin online: buy amoxil – purchase amoxicillin online
http://ciprodelivery.pro/# cipro ciprofloxacin
http://ciprodelivery.pro/# ciprofloxacin order online
paxlovid for sale: buy paxlovid online – paxlovid pharmacy
http://amoxildelivery.pro/# generic amoxicillin over the counter
amoxicillin medicine over the counter: amoxicillin 500mg price – amoxicillin 500mg without prescription
https://ciprodelivery.pro/# buy cipro online without prescription
elsababefactory https://sexdolllist.com/site/elsa-babe.html
https://paxloviddelivery.pro/# Paxlovid over the counter
where to buy amoxicillin pharmacy: amoxicillin 500 mg – amoxicillin 800 mg price
https://ciprodelivery.pro/# buy cipro online without prescription
http://doxycyclinedelivery.pro/# doxycycline generic
ciprofloxacin 500mg buy online: buy generic ciprofloxacin – buy ciprofloxacin
https://paxloviddelivery.pro/# paxlovid covid
paxlovid generic: paxlovid buy – Paxlovid buy online
https://clomiddelivery.pro/# can i get cheap clomid tablets
Respect to post author, some fantastic information
My website: analporno.club
Is it possible to get a refill on thyroid medication without a doctor’s visit cenforce 100 opinie?
buy cipro online usa: cipro – purchase cipro