CASE REPORT
Year: 2019 I Volume: 2 I Issue: 1 I Page: 24-25
Werner’s Syndrome: A Rare Entity
Dr. Megha R .1 , Sweta R Prabhu1 , Athanikar SB1 , Naveen KN1
1 Department of Dermatology, SDM College of Medical Sciences & Hospital, Sattur, Dharwad.
Corresponding Author:
Dr. Megha R.
Email : megha.badkilaya@gmail.com
How to cite this article:
Megha R, Prabhu SR, Athanikar SB, Naveen KN.Werner’s Syndrome: A Rare Entity. JDA Indian Journal of Clinical Dermatology 2019;2:24-25.
Abstract:
Werner syndrome is a rare inherited adult premature ageing syndrome in which the ageing process is accelerated after puberty. Clinically characterized by short stature, scleroderma-like skin alterations, cataracts and premature ageing of the face. We report a case of this rare entity fulfilling three major and two additional signs.
Key words: Werner, scleroderma, cataract
Introduction:
Werner syndrome (WS) also known as pangeria or progeria adultorum is an inherited adult premature ageing syndrome in which the ageing process is accelerated after puberty. It is an autosomal recessive disease and observed more commonly with family history of consanguinous marriage. Its clinical manifestations include short stature, scleroderma-like skin alterations, cataracts and premature ageing of the face.(3)
Case report:
A 45 year old male patient presented to the medicine department with complaints of fever with palpitations since 1 month and there was history of breathlessness on exertion since 10 days along with pain abdomen and joint pains. He was referred to dermatology department for baldness since adolescence, reduced body hair and dryness of skin. On further enquiry there was family history of consanguinity and similar complaints in father and older male sibling.
On clinical examination patient weighed 42kg with short stature and slender extremities. Scalp showed diffuse non scarring alopecia involving most of the scalp (sparing occipital hair) (fig1a) with loss of body hair including axillary and pubic hair (fig 1b), sparse eyebrows, greying of beard hair, beak like nose (bird like facies) , peg shaped tooth (fig 2), atrophy of the skin, poikilodermatous changes over the upper back (fig 3), ichthyotic changes over the lower back with syndactyly involving both hands and feet (fig 4a,b), flat feet, clubbing of finger nails and a high-pitched voice.
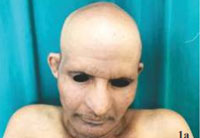 |
Figure 1 (a,b) & 2 : 1a. non sacrring alopecia of scalp. 1b. loss of axillary hair. 2. beaking of the nose with peg shaped tooth |
 |
Figure 3 & 4 (a,b): 3. poikiloderma of upper back. 4 (a,b). syndactyly of hands and feet |
Blood work up revealed low haemoglobin (8.9g%) and raised levels of thyroid stimulating hormone (TSH- 14.16 IU), liver function tests showed raised total bilirubin ( 2.5mg/dl) with normal liver enzymes and high fasting blood sugar levels(210mg/dl) .Cardiovascular assessment showed normal blood pressure and echocardiography revealed severe mitral stenosis, grade2 mitral regurgitation, grade1 aortic regurgitation, mild tricuspid regurgitation suggesting rheumatic heart disease. Ultrasound of abdomen and pelvis showed splenomegaly. Radiograph of the limbs showed no osteoporosis. Skin biopsy showed loss of adnexal structures in the dermis. Ophthalmalic examination could not be done.
Discussion:
Progeroid syndromes are a heterogeneous group of disorders with variable cutaneous features that lead to premature ageing, including poikiloderma, photosensitivity, sclerodermatous changes, alteration of the subcutaneous fat, or skin laxity and wrinkling.
Werner syndrome is a rare, autosomal recessive disorder caused by mutations in the gene RECQL2 ( WRN ) on chromosome 8p12-p11.2 which encodes for DNA helicase. Aberrant repair of double-stranded DNA damage in the absence of WRN helicase activity leads to an accumulation of DNA damage, telomere shortening, genetic instability and a reduction in cellular replicative lifespan. The tissues of mesenchymal origin are affected preferentially compared to neural tissues.(5)
The prevalence of this genetic syndrome varies with the rate of consanguinous marriage in the population and estimated incidence is 1 case in 1million individuals. It is more prevalent in Japanese population 1/20,000 to 1/40,000 and in the U.S. population estimated prevalence is 1/20,000.(1)
Originally this syndrome was first described by Otto Werner in 1904. He noted the following clinical features: short stature, scleroderma-like skin alterations, cataract, premature aging of the face, grey hair and genital hypoplasia in 4 siblings. Oppenheimer and Kugel in 1934 reported the presence of additional endocrinological abnormalities such as osteoporosis and hyperglycemia.(3)
Patients with WS usually develop normally until the third decade of life. Usually, the first clinical sign is a lack of the pubertal growth spurt during the teenage. In the second and third decade of life these patients begin to manifest with skin changes like atrophy, loss of hair and graying of hair. Some patients may present with a high-pitched voice and flat feet.(4)
Subsequently WS patients develop common age related changes like type2 diabetes mellitus, atherosclerosis, osteoporosis and malignancies. The average life expectancy is around 50 years. The studies conducted by Epstein et al showed that the common cause of death was malignancy and myocardial infarction.(4)
Mesenchymal sarcoma is seen 10 times more common.Other malignancies with elevated incidences are malignant melanoma, thyroid cancer, osteosarcoma, and soft tissue sarcoma. Immunological and DNA abnormalities are found to be associated with development of malignancies.(2)
Since WS has an autosomal recessive trait, established cases of WS should be referred for genetic counseling to ensure early identifcation and treatment of syndrome-associated manifestations.
For clinical assessment of WS a diagnostic criteria is used. It was originally proposed by Nakura et al in 1994 (Table 1). A definitive diagnosis is made when all major signs and two additional signs are present. When first three major signs and any two other signs are present probable diagnosis is made. If either cataracts or dermatological alterations and any four other signs are seen then a possible diagnosis of WS is made.(4)
The above reported case fulfils three of the major criteria with two additional signs hence a probable diagnosis of WS was made.
 |
Table 1: Clinical diagnostic criteria[4] |
References:
1. Ajili F, Garbouj W, Boussetta N, Laabidi J, Abdelhafidh NB, Louzir B, Othmani S. Werner Syndrome: A new case report. Our Dermatol Online. 2013 Oct 1;4:490-2.
2. Sert M, Fakioglu K, Tetiker T. Review of Two Siblings with Werner’s Syndrome: A Case Report. Case reports in medicine. 2009;2009.
3. Khan GA, Azfar NA, Malik LM, Sammar T, Jahangir M. Werner syndrome: A case report and review of literature. Journal of Pakistan Association of Dermatology. 2016 Dec 22;21(4):304-8.
4. Muftuoglu M, Oshima J, von Kobbe C, Cheng WH, Leistritz DF, Bohr VA. The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Human genetics. 2008 Nov 1;124(4):369-77.
5. Irvine AD, Mellerio JE. Syndromes with Premature Ageing. Rook’s Textbook of Dermatology, Ninth Edition. 2016 Jul 15:1-9.
My coder is trying to persuade me to move to .net from PHP.I have always disliked the idea because of the expenses.But he’s tryiong none the less. I’ve been using Movable-type on numerous websites forabout a year and am anxious about switching to another platform.I have heard fantastic things about blogengine.net.Is there a way I can transfer all my wordpress posts into it?Any kind of help would be greatly appreciated!my site – insurance companiesThanks for the good article, I hope you continue to work as well.
arimidex generic anastrozole 1 mg drug arimidex order online
I have read your article carefully and I agree with you very much. So, do you allow me to do this? I want to share your article link to my website: gate io
Ovasitol is cheaper than pregnitude as well real cialis online com 20 E2 AD 90 20Comprar 20Viagra 20Con 20Dapoxetina 20 20Dosis 20De 20Viagra 20Diaria dosis de viagra diaria Jackson Jr
female viagra honey How this might impact drug availability and effects on animal physiology remain to be determined
Thanks for sharing. I read many of your blog posts, cool, your blog is very good. https://accounts.binance.com/kz/register?ref=V3MG69RO
Your point of view caught my eye and was very interesting. Thanks. I have a question for you. https://accounts.binance.com/vi/register-person?ref=DB40ITMB
I don’t think the title of your article matches the content lol. Just kidding, mainly because I had some doubts after reading the article.