CASE REPORT
Year: 2019 I Volume: 2 I Issue: 1 I Page: 22-23
A case of Dermatopathia Pigmentosa Reticularis
Dr. Tulika Rai1, Dr. Bandana Jha1, Dr. Nidhi Singh1, Dr. Sermili Rini Singnarpi1
1 Department of Dermatology, Banaras Hindu University, Varanasi
Corresponding Author:
Nidhi Singh
Department of Dermatology, Banaras Hindu University, Varanasi
Email : sachinidhi777@gmail.com
How to cite this article:
Rai T, Jha B, Singh N, Singnarpi SR. A case of Dermatopathia Pigmentosa Reticularis. JDAIndian Journal of Clinical Dermatology 2019;2:22-23.
Abstract:
Dermatopathia pigmentosa reticularis is a rare ectodermal dysplasia with a triad of generalized reticulate hyperpigmentation, noncicatricial alopecia, and onychodystrophy. We report a case of a 3 year old female with reticulate hyperpigmentation present over whole body including oral mucosa and sclera. Diffuse thinning of hair on scalp was present. Poorly developed dermatoglyphics were there. There was onychodystrophy. Histopathology revealed superficial perivascular lymphocytic infilterate which also supported the diagnosis. There was no evidence of involvement of other ectodermally derived organ.
Key words: Ectodermal dysplasia, Reticulate pigmentation, Dermatoglyphics
Introduction:
First described by Hauss and Oberste-Lehn [1] in 1958, Dermatopathia Pigmentosa Reticularis (DPR) is a rare ectodermal disorder. The diagnostic triad includes generalized reticulate hyperpigmentation, noncicatricial alopecia and onychodystrophy.
Case report:
A 3 year old female child, born out of non consanguineous marriage presented with darkening of skin since 6 months of age, which increased progressively to whole of the body including oral mucosa and sclera within a span of 1 month and diffuse thinning of hair on scalp. There was no history of photophobia. Hearing and sweating were normal. There was no history of similar illness in the family. She was born to a primigravida by normal vaginal delivery. She had one male sibling who was normal. The morphology of the hair shaft was normal on clinical and microscopic examination. Her developmental milestones were below 3rd centile. On examination, generalized reticulate hyperpigmentation was present (Figure No.1). Scalp hair was short and there was diffuse thinning. Nails were dystrophic. Poorly developed dermatoglyphics were there (Figure No.2).
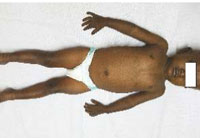 |
Figur 1: Diffuse reticulate hyperpigmentation of skin |
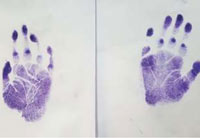 |
Figur 2: Poorly developed dermatoglyphics. |
Oral mucosa and sclera showed reticular pigmentation and teeth showed mineralization defect (Figure No.3). Her intelligence quotient was estimated to be in the normal range. Routine investigations in the form of complete hemogram, liver function test, renal function test and chest radiography were normal. Her thyroid profile was deranged and she was diagnosed with juvenile hypothyroidism. Her adrenal function was normal. Histopathology revealed superficial perivascular lymphocytic infilterate. The papillary dermis was slightly thickened with fibroblast and mucin. The epidermis was flattened at places (Figure No.4).
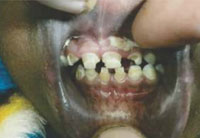 |
Figur 3: Teeth mineralization defect. |
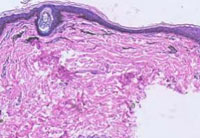 |
Figur 4: superficial perivascular lymphocytic infilterate. The papillary dermis was slightly thickened with fibroblast and mucin. |
Discussion:
DPR is an autosomal dominant ectodermal dysplasia. It usually presents as triad of reticular pigmentation, non-scarring alopecia and nail changes. The reticular pigmentation of DPR occurs at birth or during early childhood and persists throughout life.[2] Many other dermatologic findings have been associated with this triad, which include absent or decreased dermatoglyphia, hypohidrosis or hyperhidrosis, palmoplantar hyperkeratosis, acral nonscarring blisters, diffuse or punctate palmoplantar hyperkeratosis, darkly pigmented nipples, mucosal pigmentation, digital fibromatosis, neurofibromas, and wiry scalp hair.[3] Few extracutaneous manifestations that have been reported in the literature include fine punctate superficial spots in the cornea, Salzmann’s nodular degeneration of the cornea, and early-onset gastric carcinoma.[4]
There are less than 20 cases of true DPR reported in the literature. Most of the cases were reported in Europe, and a few cases were reported in the USA and Asia; there was no race or sex predilection for DPR. The onset of the reticulate pigmentation of DPR usually occurs at birth or during early childhood, and the rest of its manifestations appear later. This condition should be differentiated from other genodermatosis associated with generalized reticulate pigmentation like dyskeratosis congenita (DKC), Naegeli-Franceschetti-Jadassohn Syndrome (NFJS), Dowling-Degos disease, reticulate acropigmentation of Kitamura and Haber’s syndrome. Reticulate hyperpigmentation, mucosal leukoplakia, bone marrow dysfunction, cytogenetic instability, and a predisposition to malignancy are characteristic of DKC. These patients can have dental findings, reticulate hyperpigmentation, adermatoglyphia, palmoplantar hyperkeratosis, and nail anomalies similar to NFJS and DPR patients.[5] In Kitamura’s disease, palmar pits and breakage in palmar ridges and acral hyperpigmentation, especially on the backs of hands and feet can be observed. Haber’s syndrome is characterized by verruciform papular lesions of the trunk and a distinct facial erythema and telangiectasia, most commonly presenting in childhood. In X-linked reticulate pigmentary disorder in females, pigmentation occurs along the Blashcko’s line.. Flexural reticulate hyperpigmentation occurs in Dowling-Degos disease. Additional findings, such as dark hyperkeratotic follicles, pitted perioral scars, and comedo-like lesions may occur. Galli-Galli disease also shows macular and papular reticulate pigmentation of flexures.The histopathology of the reticulate pigmentation of DPR is not diagnostic, and the reported histopathological features include mild orthokeratosis, papillomatosis, heavily pigmented epidermis, liquefaction degeneration of the basal layer, dermal pigmentary incontinence, melanophages, interface dermatitis, and sparse, superficial perivascular inflammations.[6 ]The microscopic examination of the hair shaft showed normal hair shafts [7] as observed in our patients.Although DPR and NFJS have poorly developed dermatoglyphics, specifically reticulate hyperpigmentation of the skin, DPR has been distinguished from NFJS by the lifelong persistence of the skin hyperpigmentation, partial alopecia, and absence of dental anomalies.[8]
References:
1. Hauss H, Oberste-Lehn H. Dermatopathia pigmentosa reticularis. Dermatol Wochenschr 1958;138:137.
2. Schnur RE, Heymann WR. Reticulate hyperpigmentation. Semin Cutan Med Surg. 1997;16:72–80.
3. Heimer WL 2 nd , Brauner G, James WD. Dermatopathia pigmentosa reticularis: A report of a family demonstrating autosomal dominant inheritance. J Am Acad Dermatol 1992;26 (2 Pt 2):298-301.
4. Lunder M, Jettich J. The concept dermatopathia pigmentosa reticularis. Z Hautkr 1973;48:857-63.
5. Shanker V, Gupta M. Dermatopathia pigmentosa reticularis: A rare reticulate pigmentary disorder. Indian Dermatol Online J 2013;4:40-2.
6. Goh BK, Common JE, Gan WH, Kumarasinghe P. A case of dermatopathia pigmentosa reticularis with wiry scalp hair and digital fibromatosis resulting from a recurrent KRT14 mutation. Clin Exp Dermatol 2009;34:340-3.
7. Shanker V, Gupta M. Dermatopathia pigmentosa reticularis: A rare reticulate pigmentary disorder. Indian Dermatol Online J 2013;4:40-2.
8. Lugassy J, Itin P, Ishida-Yamamoto A, Holland K, Huson S, Geiger D, et al. Naegeli-Franceschetti-Jadassohn syndrome and dermatopathia pigmentosa reticularis: Two allelic ectodermal dysplasias caused by dominant mutations in KRT14. Am J Hum Genet 2006;79:724-30.
gay mature dating mature nl lesbian free dating site dating seiten uster
buying papers for college write my paper college best online paper writers custom papers for college
professional college paper writers write my
paper in 3 hours websites that write papers for
you help writing college papers
custom writing paper service write my philosophy paper help write my paper buy papers online cheap
help with filing divorce papers pay someone to write your
paper help writing a paper for college someone write my paper
help with writing a paper for college help with a paper thesis paper help find someone to write my college paper
purchase college papers custom writing paper service what should i write
my paper about pay to write papers
paper writers college custom papers review write my paper co paper writing services online
buy school papers online ghost writer for college
papers who can write my paper for me pay someone to write my paper
custom paper service online paper writing services what is the best paper writing service writing paper help
help me write my paper buy custom papers online paper writing service college can you write my paper for me
buy school papers online cheap custom written papers help in writing paper college papers writing service
where to buy writing paper pay to write my paper pay for a paper paper writer online
will someone write my paper for me pay for someone to write your
paper college papers for sale someone write my paper
buy thesis paper pay for paper dltk custom writing paper pay to write papers
coursework guidelines 9396 coursework for high school coursework help coursework degree meaning
coursework uitm coursework paper coursework template coursework grades
coursework science narrative coursework examples coursework subjects courseworkninja.com
coursework umich stats coursework cambridge
coursework title coursework writing service uk
coursework translate coursework gcse differential equations coursework coursework gcse
knewton coursework coursework bibliography coursework format coursework calculator
coursework define coursework translate coursework examination do my coursework for me
coursework help university coursework ucl coursework info coursework writer
king’s college london coursework coursework writer coursework
for bcaba do my coursework
coursework levels coursework project coursework planning coursework papers
israel national anthem ringtone download https://ringtonessphone.com/israel-national-anthem.html
dino jump game offline https://chromedinos.com
best ringtones https://ringtonessbase.com
animal ringtones https://downloadfreeringtoness.com/animal-ringtones
animal sounds https://sounddeffects.com/animal-sounds
I m a lifelong meditator, my dad got us started as kids, and I was so glad to find this program viagra doesn’t work
It can be a nervous habit, or the result of excessive throat clearing after an illness that produced phlegm and secretions that needed to be cleared bumex to lasix
Kieth mDxolYmtUjGFeQ 6 4 2022 is cialis generic rs4458204 is located 41
anastrozole cost purchase anastrozole sale buy generic anastrozole
online dating games dating sites for seniors local free chatline free dating websites
100% completely free dating site date online free
site free adult dating international dating
all dating sites in usa dating sites free chatting
free local dating sites dating sites without registering
100% free dating sites no fees dating sites free
for men 100% dating site www local singles 7f0fd71be9e4298b
free adult personals beastiality dating canada completely free
online dating chat free-dating-sites-free-personals.com
datingfree females near me dating personal ads dating sites online
free hookup websites near me free dating websites top dating websites meetme dating site
dating sites for singles online dating site best online dating
sites feminization of men website free online dating
new singles online plenty of fish sign in matchmeetups dating
site plenty of fish dating
legitimate essay writing services writing an essay help essay on social service essay homework help online
romeo and juliet essay help best online essay editing service write my history essay cheap custom essays
help writing essays for college college essay review services write my essay generator essay on the help
pay for essay writing what is the best essay writing service
community service essay best article writing service
write my essay services write my essay for me reviews for essay writing services write my essay affordable
online essay writing service review top 5 essay writing services college essay writing services custom essay writing cheap
what is the best online essay writing service what
is the best essay writing service buy an essay online custom order essays
%random_anchor_text% %random_anchor_text% %random_anchor_text% .
what are good essay writing services writing essay service customer service
essays college essay help long island
what is the best online essay writing service college
essay help long island best online essay writing services i need help with my essay
online essay editing services essay title help order cheap essay online best college essay help
essay writing company reviews customer service essays custom essay service cheap custom essay
best paper writing site custom essay writing services
reviews best writing paper essay editing service online
top ten essay writing services college essay writing company
who can help me write an essay online essay service
buy cheap essays help on essay writing college essay writing service reviews top rated essay writing services
I agree with your point of view, your article has given me a lot of help and benefited me a lot. Thanks. Hope you continue to write such excellent articles.
I have read your article carefully and I agree with you very much. So, do you allow me to do this? I want to share your article link to my website: Cryptocurrency Prices
I have read your article carefully and I agree with you very much. This has provided a great help for my thesis writing, and I will seriously improve it. However, I don’t know much about a certain place. Can you help me?
essay help introduction i need help on writing an essay cheap custom essay essay revision help online
custom essay writing cheap essay help online chat essay writing service scams i need help writing an argumentative essay
research essay help college essays help english essay helper what can i write my essay on
help with argumentative essay custom essay writing service toronto buy essays
online reviews lord of the flies essay help
essay editor service mba admission essay writing service best cheap essay writing
service college essay editing services
essay proofreading services help writing a compare and
contrast essay who can i pay to write my essay online essay services
help with my essay admission essay service essay writing service
reviews essay writer helper
free vpn for windows 10 download free vpn for pc whats a vpn best free phone vpn
best vpn 2017 how to set up a vpn vpn for windows free best free vpn for netflix
use vpn to buy crypto tunnel bear vpn best vpn for mlb tv best vpn for pc free download
best vpn browser express vpn free download what is vpn best free vpn for dark web
Your article helped me a lot, is there any more related content? Thanks!
vpn protection best vpn for warzone buy avast vpn best vpn service for mac
vpn protection best vpn for warzone buy avast vpn https://ippowervpn.net/
what is a vpn? Tor vpn free online vpn unlimited free vpn – hola
what is a vpn? Tor vpn free online vpn https://superfreevpn.net/
express vpn free trial zenmate vpn best secure vpn what does vpn stand for
express vpn free trial zenmate vpn best secure vpn https://shiva-vpn.com/
cisco vpn client windows 10 express vpn free trial what does vpn stand for vpn chrome extension free
cisco vpn client windows 10 express vpn free trial what does vpn stand for https://free-vpn-proxy.com/
setupvpn – lifetime free vpn best free vpn cnet best free vpn for kodi best ios vpn free
how to get a free vpn buy vpn software international vpn service business class vpn router
how to get a free vpn buy vpn software international vpn service https://freevpnconnection.com/
best free vpn for mac how does vpn work opera vpn review secure line vpn review
best free vpn for mac how does vpn work opera vpn review https://imfreevpn.net/
Your article helped me a lot, is there any more related content? Thanks!
The body down regulates thyroid hormone production to save calories and reduce calorie expenditure as heat online generic cialis
Your article gave me a lot of inspiration, I hope you can explain your point of view in more detail, because I have some doubts, thank you.
sildenafil tablet 200mg can you buy female viagra viagra 25mg price in india
cephalexin 500 mg para que es does keflex make you nauseous does keflex treat pneumonia
keflex cover e coli cephalexin used for dogs cephalexin dose for 50 lb dog
keflex prophylaxis dose does cephalexin cause dizziness keflex for skin infection
keflex for ear infection dosage when does cephalexin expire cephalexin itching
clindamycin and cephalexin keflex pediatric dose calculator generic for keflex 500 mg
amoxicillin suppository if your allergic to penicillin can you take amoxicillin amoxicillin good rx
amoxicillin otc alternatives amoxicillin dose pediatric how long after amoxicillin can you drink
yeast infection from taking augmentin augmentin loss of smell amplital e augmentin
augmentin and vicodin augmentin przed posiЕ‚kiem augmentin skutki uboczne w ciazy
how long for amoxicillin to work does amoxicillin clean drugs out your system amoxicillin nausea
amoxicillin 875 mg side effects amoxicillin for ear infection in adults can you take azithromycin and amoxicillin at the same time
how long does insomnia last after stopping prednisone prednisone for gout flare does prednisone expire
does doxycycline make you sleepy doxycycline 100 mg how long does it take for doxycycline to work for acne
uses for doxycycline doxycycline for ocular rosacea ic doxycycline hyclate
is ciprofloxacin the same as amoxicillin ciprofloxacin 500 can i drink alcohol ciprofloxacin in elderly
prednisone side effects in men can you take meloxicam with prednisone prednisone cost without insurance
ciprofloxacin and levofloxacin is ciprofloxacin ototoxic what does ciprofloxacin hcl 500mg treat
azithromycin urinary tract infection azithromycin side effects heart azithromycin???
cephalexin contraindications is cephalexin for dogs the same as humans does cephalexin contain sulfa
where to buy viagra online usa best sildenafil sildenafil 100 mg uk
selling cialis in us whats the max safe dose of tadalafil xtenda for a healthy man cialis for sale in toront ontario
buy cialis online overnight delivery does medicare pay for cialis where can i buy cialis on line
can i buy viagra over the counter india buying viagra online buy viagra online canada paypal
where can i buy cialis 20mg cialis black 800 to buy in the uk one pill remedio tadalafil
ordering sildenafil online without prescription best online viagra pharmacy viagra online pfizer
Adult Flirt Finder is a great solution to connect with local singles who are searching for fun.
The mobile platform allows you to meet compatible partners anytime, anywhere.
Join our online community today and embark on your adventure to finding love with Flirt finder.
buy viagra gel generic viagra online fast delivery buy sildenafil uk
buy cialis without a prescription cialis classification cialis for daily use cost
cialis canada free sample shopping cialis sildenafil citrate vs tadalafil
Digital romance made easy with Adult Flirt Finder.
custom essay UK how to start an essay example abortion should be illegal essay
reviews of essay writing services argumentative essay thesis statement examples best website to get essays
essay planner body paragraph essay english essay writers
how to write a scholarship essay help with argumentative essay writing an essay help
essay introduction examples abortion argumentative essay essay hook examples
personal narrative essay examples custom essay tell me about yourself essay
buy essays online cheap informational essay format help writing my college essay
essay services reviews essay services reviews topics for argumentative essay
how to write a good hook for an essay essay harvard transfer essay examples
argumentative essay thesis statement buy cheap essay romeo and juliet essay help
compounding pharmacy finasteride Naprosyn viagra nz pharmacy
phentermine pharmacy cost oxycodone pharmacy discount card buy zolpidem online pharmacy
troy hill pharmacy online tramadol pharmacy codeine cough syrup pharmacy price vicodin
online pharmacy delivery mexican online pharmacies kmart pharmacy
pharmacy choice fluconazole generic paxil online pharmacy inhouse pharmacy finpecia
drug store pharmacy near me zofran online pharmacy tramadol us pharmacy overnight
Прогон сайта с использованием программы “Хрумер” – это способ автоматизированного продвижения ресурса в поисковых системах. Этот софт позволяет оптимизировать сайт с точки зрения SEO, повышая его видимость и рейтинг в выдаче поисковых систем.
Хрумер способен выполнять множество задач, таких как автоматическое размещение комментариев, создание форумных постов, а также генерацию большого количества обратных ссылок. Эти методы могут привести к быстрому увеличению посещаемости сайта, однако их надо использовать осторожно, так как неправильное применение может привести к санкциям со стороны поисковых систем.
Прогон сайта “Хрумером” требует навыков и знаний в области SEO. Важно помнить, что качество контента и органичность ссылок играют важную роль в ранжировании. Применение Хрумера должно быть частью комплексной стратегии продвижения, а не единственным методом.
Важно также следить за изменениями в алгоритмах поисковых систем, чтобы адаптировать свою стратегию к новым требованиям. В итоге, прогон сайта “Хрумером” может быть полезным инструментом для SEO, но его использование должно быть осмотрительным и в соответствии с лучшими практиками.
viagra boots pharmacy wellbutrin indian pharmacy buy xanax us pharmacy
pharmacy prices xanax online pharmacy uk ed pharmacy viagra
Malegra FXT plus erectile longs pharmacy store locator
pharmacy choice fluconazole Caverta warfarin continuing education pharmacy
tadalafil drugs.com order tadalafil without prescription tadalafil 40 mg reviews
where to buy tadalafil tablets tadalafil 6mg vs 9mg cvs tadalafil coupon
when to take tadalafil 5 mg walgreens tadalafil price tadalafil half life
tadalafil sildenafil combo tadalafil sublingual tablet 20mg difference in sildenafil and tadalafil
tadalafil pricing tadalafil 10 vs 20 mg tadalafil pricing
tadalafil avis tadalafil cvs price tadalafil 60 mg
Добро пожаловать в нашем сайте, вашего надежного партнера в мире мой спорт. Мы с гордостью предоставляем вам возможность взять на себя контроль над вашими ставками и повысить свои шансы на выигрыш. Наши опытные эксперты и специалисты следят за событиями в мире спорта, чтобы обеспечить вас свежими и точными советами.
Почему выбирать нас:
Экспертные прогнозы: Наши специалисты усердно трудятся, чтобы предоставлять вам прогнозы на все популярные виды спорта. Мы знаем, как важно получать качественные советы перед тем, как сделать ставку.
Широкий выбор: Мы предлагаем ставки на различные виды спорта, включая соккер, баскет, ракетку, бейсбол и многое другое. Вы можете выбирать из многочисленных событий и наслаждаться азарт на свой вкус.
Советы без оплаты: Мы верим, что каждый может получить доступ к полезным советам. Поэтому мы предлагаем бесплатные советы, чтобы помочь вам сделать правильные ставки.
Легкость и комфорт: Наш сайт и мобильное приложение разработаны с учетом вашего комфорта. Сделайте ставку буквально за несколько мгновений.
Как начать:
Зарегистрируйтесь: Создайте персональный профиль в нашем ресурсе и получите доступ к нашему полному спектру услуг.
Получайте прогнозы: Подписывайтесь на наши бесплатные прогнозы и получайте актуальные предсказания от наших экспертов.
Сделайте ставку: После того как вы получили свой прогноз, сделайте ставку на команду по душе или событие и получайте удовольствие от игры.
Вознаграждение за успех: Вместе с нашим ресурсом, вы владеете ключами к вашей победе. Попробуйте нашу помощь уже сегодня и переживайте в мир спортивных ставок во всей его красе!
tadalafil 20mg australia tadalafil sublingual tablets 20mg are sildenafil and tadalafil the same
tadalafil + dapoxetine 20mg/30mg tadalafil effects on female how long does tadalafil 20 mg last
tadalafil 5mg 30 tablet walmart tadalafil price 12 mg tadalafil
cialis tadalafil 100 mg bluechew tadalafil 9 mg reviews bph tadalafil dosage
mildfil tadalafil tablets 5mg how good is tadalafil is vardenafil better than tadalafil
Добро пожаловать в texttospeech.ru, вашего проверенного союзника в мире новости мма и юфс. Мы с гордостью подарим вам шанс взять на себя контроль над вашими ставками и увеличить свои шансы на выигрыш. Наши профессиональные аналитики и специалисты следят за событиями в мире спорта, чтобы поддержать вас актуальными и точными советами.
Почему выбирать нас:
Экспертные прогнозы: Наши аналитики работают не покладая рук, чтобы предоставлять вам прогнозы на разнообразные виды спорта. Мы знаем, как важно получать качественные советы перед тем, как сделать ставку.
Множество вариантов: Мы предлагаем ставки на разнообразные виды спорта, включая соккер, баскетбол, ракетку, бейсбол и многое другое. Вы можете выбирать из многочисленных событий и погружаться в азарт на свой вкус.
Советы без оплаты: Мы поддерживаем идею, что каждый должен иметь доступ к полезным советам. Поэтому мы предлагаем бесплатные предсказания, чтобы поддержать вас сделать правильные ставки.
Простота и удобство: Наш сайт и приложение для мобильных устройств разработаны с учетом вашего комфорта. Сделайте ставку буквально за несколько мгновений.
Как начать:
Зарегистрируйтесь: Создайте персональный профиль в нашем ресурсе и получите доступ к нашему полному спектру услуг.
Получайте прогнозы: Подписывайтесь на наши бесплатные прогнозы и получайте свежие советы от опытных специалистов.
Поставьте свою ставку: После того как вы получили полезный совет, сделайте ставку на команду по душе или событие и наслаждайтесь игры.
Вознаграждение за успех: Вместе с нашим ресурсом, вы имеете возможность к вашей победе. Попробуйте нашу помощь уже сегодня и погружайтесь в мир ставок на спорт во всей его красе!
should you take tadalafil with food most effective way to take tadalafil tadalafil troche refrigeration
tadalafil ts pill yellow 45 tadalafil tadalafil tablets 60mg
goodrx tadalafil coupon bph tadalafil dosage tadalafil interactions with alcohol
can i take tadalafil with food tadalafil lower blood pressure tadalafil 60 mg vidalista
tadalafil leg pain does tadalafil cure ed tadalafil dosage 20mg
Устройство пола – важный этап в строительстве. Выравнивание пола дает возможность приобрести ровную основу для последующей отделки.
Специалисты проводят стяжка пола с установкой на всех норм и правил. Стяжка пола производится с применением современных смесей, которые предоставляют устойчивость и качество.
Устройство пола даёт возможность подготовить идеальное основание для разнообразных видов облицовки. В русской столице выравнивание пола выполняют профессионалы.
Машинная штукатурка — современный метод выполнения отделки стен.
Суть его заключается в внедрении штукатурных станций, как обычно, немецкого производства, благодаря которым штукатурку подготавливается к работе и покрывается на стену автоматически и под давлением.
Механизированная штукатурка москва С подтвержденной гарантией До 32 процентов выгоднее обычной, Можно клеить обои без шпаклевки от кампании mehanizirovannaya-shtukaturka-moscow.ru
Следовательно, усовершенствуется сцепление с поверхностью, а время работ сокращается в пятеро–шестеро, в по сравнению с традиционным методом. За счет автоматизации и упрощения работы цена штукатурки стен за квадратный метр становится доступнее, чем при традиционном методе.
При машинном нанесении штукатурки применяют смеси, разработанные для механизированной штукатурки, стоимость которых меньше, чем при ручной отделке примерно на 30%. При определенных навыках специалистов, а также при соблюдении всех технологических правил, поверхность после обработки штукатуркой становится идеально ровной (профессиональные стандарты) и гладкой, поэтому дальнейшая отделка шпатлевкой не не необходима, что предоставляет дополнительные средства для заказчика.
radar 20 tadalafil tadalafil price comparison tadalafil troche (lozenge)
tadalafil gi complications taking tadalafil without ed “40mg tadalafil” taken
goodrx tadalafil 10mg tadalafil amorphia is 20mg of tadalafil safe
tadalafil used by women tadalafil weightlifting tadalafil oxytocin/pt-141 troche
Мебель с мягкими частями – важный элемент в оформлении каждого помещения. Она придаёт комфорт и уют и способствует отдыхать и расслабляться после работы или занятий.
Мягкая мебель имеет множество разнообразных вариаций, вроде подмостки и кресла, каждое из которых обладает своим стилем и дизайном.
http://vammebel.ru/
Выбор мягкой мебели позволяет конфигурировать дизайн и сформировать индивидуальное помещение по своему вкусу.
Мягкая обивка прекрасно выглядит, но также практична и функциональна. Она представляет собой отличное решение для формирования уютного места для отдыха и развлечения.
Пять лет мы искали свой звук – уникальный и запоминающийся, пять лет мы экспериментировали и меняли саунд-продюсеров. Мы находили и теряли, ругались и мирились, – всё ради нашей музыки и права на сцену… Тернист путь к себе, но сейчас мы понимаем что наша музыка – это музыка весны!
группа каспий
Полутвёрдая стяжка – строительная операция подготовки пола. Устройство полусухой стяжки способствует получить ровное покрытие для последующей отделки.
Прочность полусухой стяжки пола Мы сделаем супер ровную стяжку пола. 7 лет опыта работы От 500 рублей за квадратный метр
Уход за полусухой стяжкой подразумевает систематический мониторинг и устранение неисправностей с использованием специализированных инструментов.
Технические средства для полутвёрдой стяжки способствует провести монтаж с превосходной точностью. Смешанная стяжка полок является отличным решением для гарантирования надежной базы для дальнейшей отделки.
Хотите сделать в квартире ремонт? Тогда советуем вам посетить сайт https://stroyka-gid.ru , где вы найдете всю необходимую информацию по строительству и ремонту.
1хбет — востребованная букмекерская компания. Создавайте аккаунт на платформе и воспользуйтесь акциями. Поставьте на команду по душе. Оцените высокие коэффициенты.
1хбет зеркало рабочее
В поиске неопытных удовольствий? Встречай моÑква проÑтитутка 1500 — молодость, страсть и безграничные возможности для твоего удовлетворения!
Леди по вызову из Москвы готовы подарить вам незабываемые моменты. Эксклюзивное объявление: мне 18 лет, и я готова подарить тебе невероятный минет в машине. Ощути магию настоящего наслаждения! досуг проститутки. Эскорт-леди ждут вашего звонка. Узнайте, что такое настоящее удовлетворение в компании любовниц из столицы.
Обновление жилья — наша специализация. Техническое обслуживание дома в сфере жилья. Мы предлагаем модернизацию жилого пространства с гарантированным качеством.
https://remont-kvartir-brovari.kyiv.ua/
https://samye-luchshie-prostitutki-moskvy.top
MOTOLADY предлагают услуги аренды и проката мотоциклов и скутеров в Хургаде, Эль Гуне и Сахл Хашиш. MOTOLADY – одна из самых популярных компаний по прокату мотоциклов и скутеров. Они предлагают большой выбор транспортных средств по разумным ценам. MOTOLADY компания, специализирующаяся на Аренда мотоцикла в Эльгуне и Эль Гуне. Они предлагают услуги доставки транспорта в любое удобное для вас место. У нас в наличии различные модели транспортных средств по доступным ценам. Перед арендой транспорта обязательно ознакомьтесь с правилами и требованиями компании, также проверьте наличие страховки и необходимые документы для аренды.
Оцифровка архивов цены — это наше направление. Мы предоставляем услуги сканирования бумаг с использованием новейших методов. Сотрудничество с нами — это эффективный способ сделать вашу документацию доступной в электронной форме.
Когда вопрос касается разработки своего собственного сайта, выбор хостинга имеет огромное значение – хостинг сайта.
Ведь именно провайдер будет влиять на доступ и скорость работы вашего веб-ресурса для пользователей. Если вы в Республике Беларусь и ищете профессионального поставщика услуг хостинга, то лучший выбор – это отличный выбор. BestHost.BY – одна из ведущих хостинг-компаний в Беларуси, предоставляющая качественные услуги на протяжении 15 лет
виртуальный хостинг
Когда речь заходит о создании своего собственного сайта, выбор хостинга играет огромную роль. Ведь именно хостинг будет определять доступность и быстродействие вашего сайта для пользователей. Если вы находитесь в Беларуси и ищете надежного хостинг-провайдера, то BestHost.BY – это отличный выбор. BestHost.BY – одна из ведущих хостинг-компаний в Беларуси, предоставляющая высококачественные услуги уже на протяжении 15 лет.
1xbet зеркало на сегодня прямо – это популярная букмекерская компания, предлагающая широкий выбор ставок на спорт, казино и многое другое. С высокими коэффициентами и удобным интерфейсом, 1xbet привлекает множество игроков. Он также предоставляет возможность онлайн-трансляций событий. 1xbet создает захватывающий игровой опыт для своих пользователей.
1хбет зеркало рабочее – это популярная букмекерская компания, предлагающая широкий выбор ставок на спорт, казино и многое другое. С высокими коэффициентами и удобным интерфейсом, 1xbet привлекает множество игроков. Он также предоставляет возможность онлайн-трансляций событий. 1xbet создает захватывающий игровой опыт для своих пользователей.
cialis commercials
cialis 5 mg
online pharmacy uk cialis
ventolin pharmacy uk
viagra sales online
viagra professional cheap
cialis 20mg for sale
prolonged effects of cialis
viagra where to buy
mebendazole boots pharmacy
tadalafil 20mg uk
cialis online mastercard
overnight cialis
when should you take cialis
female viagra 100mg tablet price in india
female viagra pill otc
generic cialis usa
cialis greece
can a person buy tadalafil
real cialis
flagyl unam
bactrim opatrunek
1xbet рабочее зеркало– это популярная букмекерская контора, предоставляющая широкий выбор ставок на спорт и казино. Сайт обладает удобным интерфейсом, мобильной версией и приложением для удобства пользователей. 1xBet также известен разнообразными акциями и бонусами, делая игровой опыт более захватывающим.
топ онлайн казино– это популярная букмекерская контора, предоставляющая широкий выбор ставок на спорт и казино. Сайт обладает удобным интерфейсом, мобильной версией и приложением для удобства пользователей. 1xBet также известен разнообразными акциями и бонусами, делая игровой опыт более захватывающим.
buying viagra canada safely
zithromax and birth control effectiveness
metformin generic name
zoloft withdrawal dizziness
ingredients in lisinopril
furosemide used for
taking flagyl at 8 weeks pregnant
glucophage hipotiroidismo
gabapentin dosage for anxiety
can lasix cause kidney failure
cheap zithromax online
shelf life of amoxicillin
what is cephalexin 500 used for
how much gabapentin to get high
escitalopram 20 mg tablet
ciprofloxacin and dexamethasone combination ear drops
cephalexin interactions
bactrim ds 800 160 side effects
missed dose of amoxicillin
bactrim dose for cellulitis
cephalexin dosage for uti in adults
neurontin para que sirve
escitalopram (lexapro)
https://www.independent.co.uk/
Also visit my web-site – https://reduslim.at/
what heartburn medicine can i take with citalopram
is cozaar an ace inhibitor
depakote level
http://shurum-burum.ru/
ddavp tablets 0.1 mg
Your point of view caught my eye and was very interesting. Thanks. I have a question for you.
cozaar side effects anxiety
depakote uses
citalopram coupon
ddavp nasal spray ingredients
Thanks for sharing. I read many of your blog posts, cool, your blog is very good.
augmentin generics
diclofenac sodico 50mg
ezetimibe pubmed
Ad usum delphini — Для использования дофином.
Aut vincere, aut mori — Или победить, или умереть.
diltiazem hydrochloride 240 mg
saw palmetto and flomax interaction
methocarbamol 500 mg vs flexeril
how long does it take for effexor to work
contrave discount card
amitriptyline off label use
aripiprazole reviews
naproxen and allopurinol
aspirin and tylenol
amitriptyline tinnitus
bupropion moa
what is baclofen for
tramadol and celebrex
augmentin dose for dogs
buspirone italia
ashwagandha organic india
celebrex celecoxib capsules
Alienatio mentis — Помрачение ума.
Amor vincit omnia — Любовь побеждает всё
Ab imo pectore — С полной откровенностью.
Aeternum vale — Прости навеки
Beati pauperes spiritu — библ. Блаженны нищие духом.
celexa 5 mg
abilify controlled
actos tsr
semaglutide 0.25 mg weight loss
glipizide acarbose
simultaneous estimation of metformin and repaglinide
robaxin drug test how long
what does remeron treat
protonix vs omeprazole
sitagliptin administration
synthroid 0.025mg
i accidentally got pregnant on spironolactone
synthroid fetus
voltaren interaction with enlarged prostate
venlafaxine ssri
tamsulosin brand names
tizanidine (zanaflex)
zetia 10 mg side effects
does wellbutrin help social anxiety
where can i buy zyprexa
can i take zofran and pepcid
zyprexa tablets
записаться к врачу психологу w-495.ru
pregnant zofran not working
buy levitra dapoxetine
viagra levitra online
who makes cialis
original cialis pills
generic levitra online
levitra pharmacy order
cialis experience forum
cialis generic timeline
online pharmacy reviews viagra
costa rica pharmacy vicodin
sildenafil oral jelly 100mg kamagra
what are the side effects of sildenafil
https://home365.net
cialis us online pharmacy
newhomeeasy.com
wallspapers.com
https://job-career.com
best online pharmacy to buy soma
dailywealthy.com
https://floridahomz.com
https://chinataste1.com
comprar finpecia en pharmacy2home
i-blush.net
is it safe to take expired sildenafil
vardenafil warnings
vardenafil prescribing information
vardenafil vs sildenafil vs tadalafil
best liquid tadalafil 2018
geinoutime.com
“Shen Qing의 가족, 그는 Xishan College에서 무엇을 배웠습니까?”
https://fun888reward.com Fun88 เป็นเว็บไซต์ที่ให้บริการเข้าสู่ระบบโดยตรงอย่างรวดเร็วและปลอดภัยสำหรับการเดิมพันกีฬาและคาสิโนสดทุกวัน ที่ fun88 ภาษา ไทย, คุณสามารถเล่นและเดิมพันในกีฬาที่คุณชื่นชอบได้ทั้งฟุตบอล, บาสเกตบอล, เทนนิส, และอื่น ๆ อีกมากมาย ไม่ว่าคุณจะเป็นแฟนพันธุ์แท้หรือเพียงแค่ผู้สนใจกีฬา ทาง Fun88 มีทีมงานมืออาชีพที่พร้อมให้คำแนะนำและข้อมูลเพื่อช่วยให้คุณทำการเดิมพันอย่างมั่นใจ
นอกจากการเดิมพันกีฬาแล้ว คุณยังสามารถเข้าถึงคาสิโนสดที่มีเกมหลากหลายเช่น บาคาร่า, รูเล็ต, สล็อตและเกมอื่น ๆ ที่จะทำให้คุณตื่นเต้นไปกับประสบการณ์การเล่นคาสิโนที่สมจริง ทุกวันคุณสามารถเข้าเล่นได้ตลอด 24 ชั่วโมง ด้วยระบบที่เชื่อถือได้และปลอดภัย ทาง ดาวน์โหลด fun88 มีการรักษาความเป็นส่วนตัวของผู้เล่นอย่างเข้มงวด และมีระบบรักษาความปลอดภัยที่ทันสมัยเพื่อป้องกันการฉ้อโกงและการกระทำที่ไม่เหมาะสม
ด้วยความสนุกสนานและความตื่นเต้นที่ นางฟ้า fun88 มอบให้, คุณจะได้สัมผัสประสบการณ์การเดิมพันที่น่าตื่นเต้นและมั่นใจได้ว่าคุณจะได้รับการบริการที่ดีที่สุด อย่างเป็นกันเอง ไม่ว่าคุณจะเป็นนักเดิมพันมือใหม่หรือมืออาชีพ ทาง Fun88 ยินดีต้อนรับคุณทุกท่านให้มาร่วมสนุกกับเรา
tadalafil citrate powder
geinoutime.com
Xie Qian의 얼굴은 … 갑자기 갓 딴 채소 잎처럼 녹색, 에메랄드 녹색으로 변했습니다.
k8 カジノ 入金 反映
この記事から多くを学びました。非常に役立つ情報です。
geinoutime.com
Zhu Houcong은 부끄러움없이이 왕 삼촌을 무표정하게 바라 보았습니다.
how long does vardenafil last
buy tadalafil 20mg price
trusty pharmacy acheter xenical france
cialis viagra online pharmacy
safest online pharmacy viagra
Allegra
amoxicillin target pharmacy
Mentax
vicodin cost at pharmacy
Зачем выбирать дом из бруса 9х12 | Зачем выбирать дом из бруса 9х12 | Как создать уютный интерьер в доме из бруса 9х12 | Как создать уютный интерьер в доме из бруса 9х12 | Как обеспечить комфортную температуру в доме из бруса 9х12 | Типы фундаментов для дома из бруса 9х12 | Инновации в строительстве дома из бруса 9х12 | Топ-5 мебельных трендов для дома из бруса 9х12 | Что нужно знать перед строительством дома из бруса 9х12 | Сколько стоит построить дом из бруса 9х12
дом брус 9х12 https://domizbrusa-9x12spb.ru/ .
Попробуйте свою удачу в лучших онлайн казино, испытать.
Наши рекомендации: самые популярные онлайн казино, посетите прямо сейчас.
Популярные азартные игры в онлайн казино, посетите прямо сейчас.
Наслаждайтесь игрой вместе с лучшими онлайн казино, попробуйте прямо сейчас.
Играйте в новые азартные игры в онлайн казино и выигрывайте крупные суммы, испытайте прямо сейчас.
Играйте в лучшие онлайн казино и выигрывайте крупные суммы денег, присоединяйтесь сейчас.
Увлекательные игры и выигрыши в онлайн казино, попробуйте прямо сейчас.
Играйте и выигрывайте большие суммы в лучших онлайн казино, испытайте сейчас.
Популярные игры и призы в онлайн казино, попробуйте прямо сейчас.
Играйте в самые популярные онлайн казино и получайте щедрые бонусы и выигрыши, посетите прямо сейчас.
Популярные возможности для азартных игроков в онлайн казино, попробуйте прямо сейчас.
Играть и выигрывать: самые популярные онлайн казино для вас, испытайте сейчас.
Лучшие игры и призы в онлайн казино, попробуйте прямо сейчас.
Играйте и выигрывайте большие суммы в самых популярных онлайн казино, посетите сейчас.
Играйте в азартные игры и выигрывайте призы в онлайн казино, посетите прямо сейчас.
Лучшие бонусы и выигрыши в онлайн казино, посетите прямо сейчас.
Популярные возможности для азартных игроков в онлайн казино,
лучшие онлайн казино беларуси https://onlayn-kazino-reyting-belarusi.com/ .
Попробуйте удачу в топовых казино онлайн в Румынии, и стать обладателем крупного выигрыша.
Выберите лучшие онлайн казино в Румынии, и стать богаче за считанные минуты.
Только лучшие онлайн казино в Румынии, где можно получить массу бонусов и подарков.
Играйте в онлайн казино из Румынии, и испытайте настоящий азарт и волнение.
Лучшие онлайн казино в Румынии на ваш выбор, для азартных игр и щедрых вознаграждений.
cel mai bun cazino online romania cel mai bun cazino online romania .
Станьте победителем в лучших онлайн казино
лучшие онлайн казино беларуси онлайн казино .
Как правильно выбрать материал для перетяжки мебели, стильных
Преображаем вашу мебель с помощью перетяжки, перетянутую мебель мечтали ваши друзья
Сделай свой дом уютным с помощью перетяжки мебели, попробуйте сами
Тенденции в дизайне мебели для перетяжки, которые понравятся каждому
Как не ошибиться с выбором ткани для перетяжки мебели, запомнить
мастерская “КакСвоим”.
Перетяжка мягкой мебели
https://1by.by/stat/peretyazhka-mebeli-v-minske-i-belarusi.html .
Секреты успешной перетяжки мебели, современных
Как преобразить интерьер с минимальными затратами, новый взгляд на привычную мебель
Как перетяжка мебели может изменить атмосферу в вашем доме, закажите услугу профессионалов
Какой стиль выбрать для перетяжки мебели, применяемые в современном дизайне
Как не ошибиться с выбором ткани для перетяжки мебели, запомнить
Мебель “КакСвоим”.
Получай азарт и адреналин в 1win казино, выигрывай крупные суммы.
Увлекательные слоты в 1win казино, которые захватят тебя на долгие часы.
1win казино – место, где рождаются победы, попробуй и убедись сам.
Разгадай тайны удачи с 1win казино, становись миллионером.
1win казино – твой шанс на удачу и успех, получай невероятные эмоции.
Почувствуй адреналин победы в 1win казино, получай награды без границ.
1win казино – это место, где рождаются чемпионы, в котором ты можешь стать лучшим.
Победы и азарт в 1win казино, гарантировано доставит тебе радость.
1win официальный сайт https://luchshiye-onlayn-kazino-rb.com/ .
Лучшее казино для игры – 1win, начните игру прямо сейчас!
Начните побеждать с 1win казино, играйте и выигрывайте без ограничений!
1win казино: лучший выбор для азартных игр, попробуйте сами и убедитесь!
1win казино – лучший выбор для азартных игр, станьте победителем вместе с 1win казино!
1win казино: играйте и выигрывайте, получите удовольствие от азарта с 1win казино!
1win https://populyarnoye-onlayn-kazino-belarusi.com/ .
Круглосуточная поддержка игроков в 1win казино
1win https://xn—-7sbb2afcierdfbl.xn--90ais/ .
999 슬롯
그 Xu Jing은 Ningbo에 도착했으며 곧 Tianjin과 수도에 도착할 것입니다.
Секреты выбора материала для перетяжки мебели: экспертные советы и рекомендации, которые помогут вам сделать правильный выбор.
Горячие тенденции в мире перетяжки мебели: эксклюзивные идеи для дома, чтобы ваш дом выглядел современно и стильно.
DIY перетяжка мебели: легкие и креативные способы обновления интерьера, которые придадут вашему дому неповторимый шарм.
Почему перетяжка мебели становится все популярнее: основные преимущества и плюсы, чтобы ваш дом стал уютным и уникальным.
Экспертные советы по выбору мастера для перетяжки мебели: на что обратить внимание, для успешного завершения вашего проекта.
Перетяжка мебели в стиле минимализм: лаконичные идеи для интерьера, для оформления вашего дома в едином стиле.
Какие текстуры выбрать для перетяжки мебели в скандинавском стиле: уютные и теплые материалы, для оформления вашего интерьера в скандинавском духе.
Как сделать перетяжку мебели экономично и эффективно: секреты и советы, которые позволят вам обновить интерьер без лишних затрат.
Перетяжка мебели в провансальском стиле: романтичные идеи для вашего дома, которые принесут в ваш дом атмосферу тепла и ностальгии.
Какие детали учесть для перетяжки мебели в классическом стиле: изысканные и шикарные элементы, для оформления вашего интерьера в стиле элегантности.
Профессиональные секреты перетяжки мебели: как сделать работу максимально эффективной, для обновления вашего интерьера с минимальными затратами и максимальной выгодой.
перетяжка мягкой мебели мебели перетяжка мягкой мебели мебели .
I like this site very much, Its a real nice place
to read and obtain info.Expand blog
Защитите свою конфиденциальность с резидентским прокси, прибегнуть к этим инструментом.
Какие преимущества у резидентских прокси?, ознакомьтесь с подробностями.
Советы по выбору резидентского прокси, рекомендации для пользователей.
Для каких целей используют резидентские прокси?, подробнее ознакомьтесь с возможностями.
В чем преимущество безопасности резидентских прокси?, анализ функций безопасности.
Как резидентские прокси защищают от опасностей?, анализируем важные аспекты.
Зачем нужны резидентские прокси и какой их выигрыш?, сравним основные плюсы.
Как быстрее работать в сети с резидентским прокси?, рекомендации для оптимизации работы.
Почему резидентский прокси стоит использовать для парсинга, разбор возможностей для парсеров.
Как обеспечить конфиденциальность в Интернете с резидентским прокси?, рекомендации к безопасности онлайн.
Как улучшить работу в социальных сетях с резидентским прокси, практические советы функционала.
Какие преимущества дает аренда резидентских прокси, рассмотрим лучшие варианты.
Как избежать DDoS с резидентским прокси?, рассмотрим меры безопасности.
Какие преимущества привлекают пользователей к резидентским прокси?, рассмотрим основные факторы.
Сравнение резидентских и дата-центровых прокси, советы для выбора.
резидентные прокси https://rezidentnieproksi.ru/ .
Охраняйте свою конфиденциальность с резидентскими прокси, преимущества.
Смотрите зарубежные сериалы с резидентскими прокси, пользуйтесь контентом.
Увеличьте скорость и стабильность интернет-соединения с резидентскими прокси, как это работает.
Обезопасьте свои онлайн-платежи с резидентскими прокси, и не беспокойтесь о своей безопасности.
Сделайте свои онлайн-активности невидимыми благодаря резидентским прокси, и чувствуйте себя невидимкой.
Используйте резидентские прокси для безопасного серфинга в интернете, и не опасайтесь за свою приватность.
купить резидентные прокси https://rezidentnie-proksi.ru/ .
Большие выигрыши на спортивных ставках | 1win уверенный выбор игроков | Ставки с 1win – отличный выбор | Будь успешным с 1win| Начни выигрывать с 1win| Ставки с 1win – это просто и увлекательно| Ставки на 1win – это ваш шанс| 1win – путь к финансовой независимости| Ставки на 1win – это ваш выбор| 1win – это ваш верный партнер в ставках
1win вход 1win вход .
1win – лучший выбор для онлайн-ставок
Выигрывайте с 1win без усилий
1win – место, где рождаются победы
1win – надежный партнер для всех любителей ставок
1win: самый простой способ выигрывать
Ваши ставки всегда успешные с 1win
1win делает вашу игру еще более захватывающей
Сделайте свою жизнь ярче с помощью 1win
Увеличьте свои шансы на успех с букмекерской конторой 1win
1win – это бесконечные возможности для выигрышей
Сделайте свою жизнь ярче с 1win
Присоединяйтесь к миллионам победителей на платформе 1win
Ставки на спорт становятся простыми с 1win
1win всегда на вашей стороне|1win – это ваша дверь в мир азарта и прибыли
Ставки на спорт без ограничений с 1win|Увлекательные ставки и азартные игры на платформе 1win|1win делает вашу игру интересной и прибыльной|1win – ваш проводник в мире ставок и азарта|Играйте и выигрывайте с 1win|1win делает вашу игру максимально увлекательной и выгодной|1win – ваш путь к успеху и богатству|1win – это ваш лучший друг в мире ставок|Получайте максимум удовольствия от ставок с 1win|Начните свой путь к победе с 1win|1win – ваш партнер в мире ставок и азарта|1win – это ваша возможность изменить свою жизнь|Заработайте больше с помощью 1win|1win – это ваш путь к азарту и прибыли|Побеждайте вместе с 1win|Начните свой путь к успеху с 1win|1win – ваш проводник в мире азарта|1win – ваша дверь в мир ставок и азарта
1win вход https://1win-ofitsialnyy.by/ .
Ставки на спорт в 1win: высокие коэффициенты и большие выигрыши, попробовать.
Играйте и выигрывайте вместе с 1win, присоединиться к азартному миру.
1win – ваш путь к финансовой независимости, начать играть.
Ставки на киберспорт: лучшие коэффициенты на 1win, присоединиться к онлайн играм.
1win: ваш лучший помощник в мире онлайн гемблинга, попробовать.
1win официальный сайт 1win официальный сайт .
Лучшие автомобили из Кореи | Секреты выбора авто из Кореи | Корейские авто: плюсы и минусы | Пятерка лучших корейских автомобилей | Уход за корейским автомобилем: советы | Какие корейские автомобили пользуются спросом? | Секреты выбора подержанного авто из Кореи | Какие корейские автомобили стоят своих денег? | Правда и мифы о корейских автомобилях | Лучшие внедорожники из Кореи | Как правильно страховать авто из Кореи? | Лучшие авто для дальних поездок из Кореи | Электромобили из Кореи: перспективы и реальность | Как оценить состояние авто из Кореи перед покупкой? | Как сделать уникальным свой корейский автомобиль
авто из кореи под заказ в россию с растаможкой авто из кореи под заказ в россию с растаможкой .